- Passive pulmonary venous hypertension and chronic increases in capillary wedge pressure (PCWP) up to 18-20 mmHg lead to extravascular interstitial stasis with impaired gas exchange and dyspnoea. Intrapulmonary oedema occurs when Pcap exceeds 20 mmHg and alveolar oedema above 25 mmHg.
- Pulmonary venous pressure chronically maintained above 20 mmHg (post-capillary pulmonary hypertension) induces a reactive increase in pulmonary arterial resistance (PAR) of a myogenic and neurovegetative nature. As intimal fibroelastosis progresses, precapillary pulmonary arterial hypertension (PHT) develops, which is disproportionate to venous hypertension. This precapillary PHT is probably adaptive: it reduces capillary blood flow and limits the risk of pulmonary oedema.
- Chronic lesions of interstitial pulmonary fibrosis reduce lung compliance and increase work of breathing, which accounts for up to 25% of total VO2. Tidal volume and vital capacity are reduced. V/Q ratios are altered and PaO2 falls as in a restrictive type of pathology. Hypoxia and hypercapnia have a vasoconstrictive effect on the pulmonary arteries, creating a vicious circle.
LVH and lack of ventricular compliance increase the pressure in the RA, which gradually dilates. Normally, atrial contraction temporarily increases LV end-diastolic pressure to optimise tension in the thick ventricular wall without the mean atrial pressure reaching levels that would cause pulmonary venous hypertension. In the absence of sinus rhythm, the mean atrial pressure required to fill the LV is much higher and the protective effect on the pulmonary circulation is lost.
Pulmonary hypertension (mean resting PAP > 25 mmHg, PAR > 240 dynes-s-cm-5) increases LV afterload [3]. In the absence of compensatory hypertrophy, the maximum systolic pressure that a normal LV can develop is 50 mmHg. PHT leads to a cascade of consequences: right-sided hypertrophy (RVH) and dilatation of the RV, tricuspid insufficiency, dilatation of the RA and systemic venous stasis. As the RV becomes enlarged, it increasingly resembles the LV and its flow becomes dependent on its preload. LV overload in turn affects left ventricular filling in several ways: 1) venous return to the LV is reduced because pulmonary flow is restricted, 2) the interventricular septum bulges into the left cavity and limits its diastolic expansion (Bernheim effect), 3) tricuspid insufficiency due to right-sided dilatation causes the interatrial septum to bulge into the LV. Coronary perfusion of the RV, which is systolic-diastolic, is compromised when its systolic intracavity pressure approaches the mean aortic pressure; in the event of failure, it must be maintained by infusion of vasoconstrictors (noradrenaline) or by intra-aortic counterpulsation [2].
Even when pulmonary hypertension is fixed, its reactive vasospastic component is important and responds to the usual pulmonary vasodilator measures: hyperventilation, alkalosis, reduction of sympathetic load and pulmonary vasodilators. The treatment of pulmonary hypertension is described in Table 12.11 (see Chapter 12, Pulmonary hypertension, Treatment). A number of vasoactive substances are released into the circulation during bypass surgery: TNF, interleukins, C3a complement, etc. They cause vasodilatation of the blood vessels. They cause systemic vasodilatation and pulmonary vasoconstriction, which occur when the heart is stressed and are easily aggravated by the administration of protamine. Aortic or mitral valve repair or replacement immediately reduces pulmonary venous pressure (postcapillary hypertension) but does not correct precapillary arterial PHT, which remains elevated for several weeks. Intimal fibroelastosis persists in the long term.
Clinical prognosis and the risk of intraoperative haemodynamic failure depend more on RV function than on the absolute value of PAP (see Figure 12.7B). Indeed, a high PAP means that the RV is capable of generating it, whereas a moderate PAP reflects a modestly increased PAR or RV dysfunction. A pulmonary hypertensive crisis may lead to right ventricular decompensation.
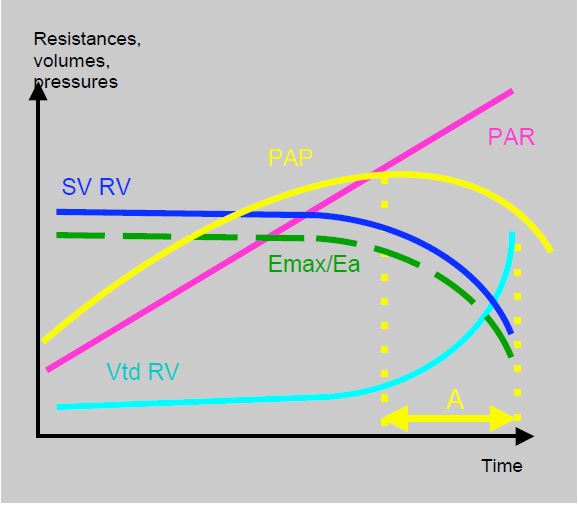
Figure 12.7B: Coupling between RV and pulmonary circulation. Diagram showing the relationship between pulmonary artery resistance (PAR), LV stroke volume (LVSV), right ventricular volume (RVdV), pulmonary artery pressure (PAP) and the Emax/Ea ratio. PAH is a function of the ability of the RV to generate chronically high pulmonary pressures. When the RV fails, PAP tends to fall while the haemodynamic situation deteriorates and SAR continues to rise. In time period A (yellow), the measured PAP is lower than before, not because of an improvement in the situation, but because of a decline in RV function [5].
For the purposes of clinical management, it is important to distinguish between three different situations, all of which can lead to right ventricular failure.
- Infarction or primary failure of the RV: the priority is to maintain sufficient coronary perfusion pressure (systemic vasoconstrictor) and optimise preload (CVP 10-15 mmHg) to obtain the best flow. Contractility is improved by inodilators (dobutamine, milrinone, levosimendan). Right-sided infarction is more common in RVH [4].
- Excessive afterload: Acute failure, e.g. due to pulmonary embolism, leading to congestive failure of the RV. The priority is to reduce right-sided preload (nitroglycerine, reverse Trendelenburg), support right-sided haemodynamics (dobutamine, milrinone ± epinephrine, norepinephrine, levosimendan) and avoid worsening of PAR.
- Pulmonary hypertension flares up in chronic PHT with right ventricular hypertrophy. The priority is to reduce PAR; preload must remain high because the hypertrophied LV operates on a Starling curve similar to that of the LV. In contrast to the previous two situations, positive pressure ventilation is well tolerated in this case because the LV afterload is already chronically high.
Specific management of right-sided decompensation is given in Table 12.6.
| Pulmonary Arterial Hypertension (PAH) |
| Left-sided congestion due to mitral or aortic valve disease leads to chronic elevation of PAR and pulmonary venous pressure (postcapillary PHT) with interstitial and possibly alveolar oedema. Venous PHT results in a reflex increase in PAR and precapillary PHT.
PHT (mean resting PAP > 25 mmHg, HBP > 240 dyne-s-cm-5 ) causes dilatation and hypertrophy of the RV (RVH). The more it hypertrophies, the more the RV functions similarly to the LV (preload-dependent flow, resistance to afterload). The clinical prognosis depends more on the function of the RV than on the absolute value of PAP. |
© CHASSOT PG, BETTEX D, August 2011, last update November 2019
References
- GUAZZI M, BORLAUG BA. Pulmonary hypertension due to left heart disease. Circulation 2012; 126:975-90
- HADDAD F, DOYLE R, MURPHY DJ. HUNT SA. Right ventricular function in cardiovascular disease, Part II. Pathophysiology, clinical importance and management of right ventricular failure. Circulation 2008; 117:1717-31
- HOEPER MM, BOGAARD HJ, CONDLIFFE R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D42-50
- KINCH JW, RYAN TJ. Right ventricular infarction. N Engl J Med 1994; 330:1211-7
- VONK NOORDERGRAAF A, et al. The relationship between the right ventricle and its load in pulmonary hypertension. J Am Coll Cardiol 2017; 69:236-43