Tetralogy of Fallot (TOF) accounts for 5-10% of congenital heart diseases. It is the most common cyanotic heart disease. Over 95% of children who benefit from surgery reach adulthood [5]. TOF is caused by a conotruncal anomaly with an anterior-right shift of the conal septum and underdevelopment of the right infundibulum. The embryonic parietal band is misaligned and abnormally positioned resulting in obstruction of the right ventricular outflow tract (RVOT) and a wide subaortic VSD [9,14]. The malformation combines four anomalies (Figure 14.56 and Figure 14.57).
- A subaortic VSD (membranous septum) (Video);
- An aorta overridling both ventricles (the aortic valve is competent) (Video);
- A stenosis of the right ventricular outflow tract and the pulmonary valve, which is restrictive, hypoplastic and bicuspid, combined with varying degrees of underdevelopment of the pulmonary artery and its branches (Video);
- A RV hypertrophy.
Video: Membranous VSD located under the aortic valve, and typical of tetralogy of Fallot.
Video: Long-axis view of the large aortic valve overriding the membranous VSD in a case of tetralogy of Fallot; the right ventricle is severely hypertrophied.
Video: Long-axis view of the right ventricular outflow tract, pulmonary valve and pulmonary artery in a case of tetralogy of Fallot; RVOT presents a massive concentric hypertrophy with a tiny lumen; pulmonary valve and PA are severely hypotrophied.
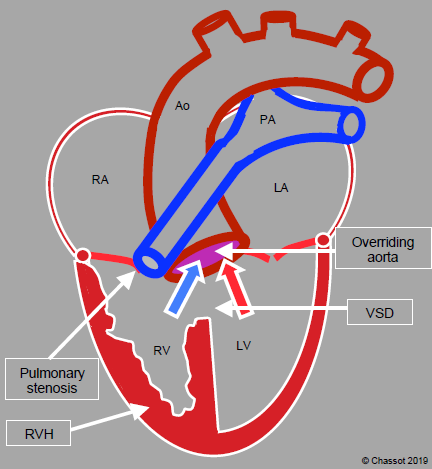
Figure 14.56: Diagram of tetralogy of Fallot. The aortic valve overrides a wide VSD. The RV is hypertrophied (RVH), although the largest diameter of its chamber is not visible in this cross-sectional plane. The pulmonary valve and pulmonary artery are stenosed.
In some cases, an ASD is added to the other lesions, which is described as pentalogy. In 10% of cases, a coronary malformation (e.g. circumflex artery originating from the right coronary artery) causes a vessel to cross the anterior face of the RVOT. Several other malformations may be associated with TOF: stenosis of the distal trunks of the PA, multiple VSDs, dextroposition of the aortic arch, pulmonary agenesis, major aortopulmonary collateral arteries (MAPCA, see below). There is a whole range of lesions similar to those of the TOF. Ten percent of all congenital heart disease patients experience some degree of pulmonary stenosis. Their anatomy is similar to Fallot cases, although without a VSD or displacement of the aorta. They are not cyanotic at rest. Tetralogy cases with minor pulmonary stenosis enabling normal oxygenation and a Qp/Qs ratio close to 1/1 are sometimes described as pink Fallot. At the other end of the spectrum, complete pulmonary atresia causes deep cyanosis.
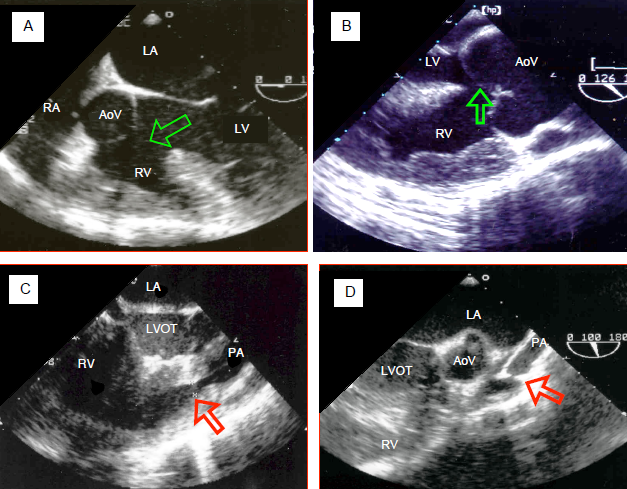
Figure 14.57: Transesophageal echocardiographic images of tetralogy of Fallot. A: 5-chamber view at 0° of the aorta opening in a overriding position over both ventricles. The arrow shows the VSD. The aortic valve receives blood from the LV and RV. The RV is hypertrophied. B: long-axis view at 120° of the aortic root. The valve overrides the VSD (arrow). The RV wall is > 1.2 cm thick and its trabeculations are hypertrophied. C: pulmonary muscular stenosis in the right outflow tract. Diameter 0.44 cm (arrow). The pulmonary valve and artery are hypoplastic. D: atresia of the pulmonary valve, whose opening is minuscule (arrow) followed by a hypoplastic and tubular pulmonary artery.
While pulmonary valve stenosis is fixed as it is caused by hypoplasia, RVOT stenosis is dynamic as this part is entirely muscular. Any increase in contractility or reduction in preload reduces its diameter, intensifies stenosis and impedes pulmonary blood flow. Sympathetic stimulation exacerbates cyanosis (blue spells), and therefore children are often administered β-blockers. This infundibular spasm gives rise to cyanotic episodes with hyperventilation requiring immediate treatment [10].
- Oxygen: FiO2 1.0;
- Squatting position, compression of the femoral arteries (increasing SVR), phenylephrine (5-10 mcg/kg);
- Morphine (0.05-1.0 mg/kg);
- Fluid administration;
- Esmolol (0.5 mg/kg).
The spontaneous development of aortopulmonary collaterals during growth enables some children with TOF to grow up normally due to the pulmonary flow supplied by these L-to-R shunts (Figure 14.58). However, this is a delicate balance since pulmonary flow is dependent on systemic flow – it increases if SVR rises but falls in the event of hypotension. If the collaterals are small, PAP remains low and the child becomes cyanotic with the slightest exertion. In contrast, if the collaterals are large, the child risks developing pulmonary hypertension. Moreover, the collaterals impose volume overload on the LV, which must provide both the systemic and pulmonary flow.
Children with TOF are cyanotic because the aorta receives a mixture of venous blood from the RV and arterialised blood from the LV, and because pulmonary flow is insufficient. Pulmonary flow and pressure are low due to pulmonary stenosis, which protects the lungs from hypertension. If systemic vascular resistance (SVR) rises, pulmonary flow rises, the R-to-L shunt decreases, and cyanosis is reduced. SaO2 directly reflects pulmonary flow. Since the RV is subjected to systemic pressure, it becomes progressively dysfunctional.
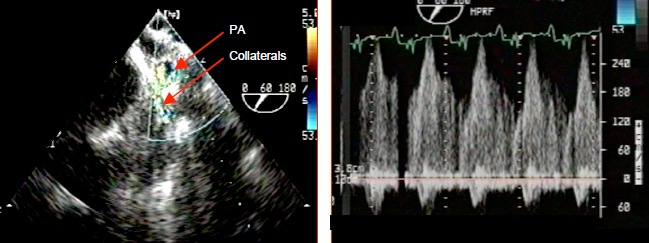
Figure 14.58: Collateral flow in a case of tetralogy of Fallot. The flow is visible between the large collateral originating from the descending aorta and a branch of the pulmonary artery (PA). It is a continuous systolic and diastolic flow, since systemic pressure is constantly higher than pulmonary pressure. Diastolic arterial pressure is low as the vessels continuously direct flow into the low-pressure pulmonary vascular bed. It rises if pulmonary hypertension takes hold. If sufficient in number, these collaterals are able to ensure normal pulmonary flow at rest.
Surgical treatment
In the nineteen seventies, two-stage surgery was generally performed on these patients. An L-to-R shunt was first performed to increase pulmonary blood flow: Blalock-Taussig shunt (subclavian artery → right or left pulmonary artery), Potts shunt (descending aorta → left PA) or Waterston shunt (ascending aorta → right PA). The shunt’s size determined its flow rate. This initial neonatal surgical procedure was followed by total correction between the ages of 5 and 10 years. Unfortunately, this strategy maintained hypoxia and pressure overload on the RV for too many years. It also caused deformity of the pulmonary arteries and hampered their normal development.
Total correction is currently performed between the ages of 2 and 10 months, which avoids neonatal surgery while also eliminating cyanosis and RVH sufficiently early, normalising pulmonary flow, and encouraging growth of the pulmonary vascular tree [4,11]. Surgery consists of separating the systemic and pulmonary circulations and removing the blockage from the pulmonary artery root (Figure 14.59) [6].
Children with TOF are cyanotic because the aorta receives a mixture of venous blood from the RV and arterialised blood from the LV, and because pulmonary flow is insufficient. Pulmonary flow and pressure are low due to pulmonary stenosis, which protects the lungs from hypertension. If systemic vascular resistance (SVR) rises, pulmonary flow rises, the R-to-L shunt decreases, and cyanosis is reduced. SaO2 directly reflects pulmonary flow. Since the RV is subjected to systemic pressure, it becomes progressively dysfunctional.
Figure 14.58: Collateral flow in a case of tetralogy of Fallot. The flow is visible between the large collateral originating from the descending aorta and a branch of the pulmonary artery (PA). It is a continuous systolic and diastolic flow, since systemic pressure is constantly higher than pulmonary pressure. Diastolic arterial pressure is low as the vessels continuously direct flow into the low-pressure pulmonary vascular bed. It rises if pulmonary hypertension takes hold. If sufficient in number, these collaterals are able to ensure normal pulmonary flow at rest.
Surgical treatment
In the nineteen seventies, two-stage surgery was generally performed on these patients. An L-to-R shunt was first performed to increase pulmonary blood flow: Blalock-Taussig shunt (subclavian artery → right or left pulmonary artery), Potts shunt (descending aorta → left PA) or Waterston shunt (ascending aorta → right PA). The shunt’s size determined its flow rate. This initial neonatal surgical procedure was followed by total correction between the ages of 5 and 10 years. Unfortunately, this strategy maintained hypoxia and pressure overload on the RV for too many years. It also caused deformity of the pulmonary arteries and hampered their normal development.
Total correction is currently performed between the ages of 2 and 10 months, which avoids neonatal surgery while also eliminating cyanosis and RVH sufficiently early, normalising pulmonary flow, and encouraging growth of the pulmonary vascular tree [4,11]. Surgery consists of separating the systemic and pulmonary circulations and removing the blockage from the pulmonary artery root (Figure 14.59) [6].
- Patch to close the VSD or direct closure with realignment of the outflow tract (trans-tricuspid route via the RA to avoid a ventriculotomy);
- Right outflow tract widened by infundibular resection and enlargement of the pulmonary valve by valvulotomy or commissurotomy, sometimes with a transannular patch;
- Valved conduit between the RV and PA if: the patient's anatomy is not suitable for correction, the pulmonary valve is atretic, or a coronary artery following an anomalous route crosses the anterior face of the RVOT.
The VSD is approached through a transatrial and trans-tricuspid route. The pulmonary valve is accessed via the PA. Avoiding a ventricular incision improves function and limits the risk of arrhythmia [12]. Widening of the RVOT entails a risk of pulmonary insufficiency if the patch is too large or of persistent stenosis if it is too restrictive. Correction is therefore based on a compromise between minimal regurgitation and minimal residual gradient. The maximum accepted gradient is 20-40 mmHg [17]. A flow rate of over 3 m/s, a gradient of over 40 mmHg or a RVsystP/LVsystP ratio > 0.5 are considered excessive. When resecting a dynamic hypertrophy of the outflow tract, the residual gradient may be functional rather than anatomical. It is reduced by rebalancing sympathetic tone (beta blockade), loading conditions, and Hb levels. Following total correction, SaO2 must be > 98%. The surgical mortality rate for total correction performed at a young age is < 2% and the survival rate is > 95% at 36 years [1,5]. However, many children require a second operation in adulthood due to pulmonary insufficiency or, less commonly, stenosis, or to implant a larger conduit. Only 10-15% of patients who undergo delayed primary correction during adolescence require subsequent reoperation. However, these children are the rare cases who have survived with their malformation for some fifteen years.
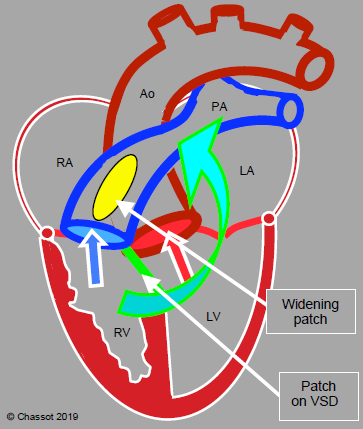
Figure 14.59: Surgical correction of tetralogy of Fallot. The VSD patch divides the flows by guiding systemic venous blood to the PA and arterialised blood to the aorta. A patch enlarging the RVOT and pulmonary valve removes the pulmonary and subpulmonary stenosis. The arrow shows the point at which a valved conduit may be inserted between the RV and PA if a patch is not feasible.
Unfortunately, correction may allow a number of major sequelae [7,12,15].
Figure 14.59: Surgical correction of tetralogy of Fallot. The VSD patch divides the flows by guiding systemic venous blood to the PA and arterialised blood to the aorta. A patch enlarging the RVOT and pulmonary valve removes the pulmonary and subpulmonary stenosis. The arrow shows the point at which a valved conduit may be inserted between the RV and PA if a patch is not feasible.
Unfortunately, correction may allow a number of major sequelae [7,12,15].
- Arrhythmias in > 50% of cases: right bundle branch blocks or bifascicular blocks (60-100%), premature ventricular contraction, AF, sudden death (1-5%). The incidence of arrhythmia can be reduced by avoiding a transventricular approach.
- Residual VSD (10-20%).
- Pulmonary insufficiency (PI). The degree of postoperative PI is the most significant prognostic factor for subsequent right ventricular function. If PI is severe (regurgitation fraction > 25%), it progressively leads to RV dilation through volume overload, which is directly linked to the incidence of ventricular arrhythmias and sudden deaths. The valve must be replaced sufficiently early to prevent right-sided complications [3]. Any increase in RV afterload, raised PVR or increased thoracic pressure intensifies regurgitation.
- Residual stenosis of the pulmonary conduit or RVOT. RV:LV pressure ratio > 0.6 is poorly tolerated in the long term and the RV fails. Correction may be performed by catheterisation: balloon dilation or percutaneous implantation of a bioprosthesis (Melody™, Sapien™) if the implantation area is sufficiently rigid (previous conduit, fibrosis) [8].
- Right ventricular failure due to volume or pressure overload occurring in adulthood. Surgical correction will not ensure adequate functional recovery of the RV if its size is > 150 mL/m2 [16]. However, it has not been demonstrated that systolic function can be normalised by reoperation, even if this is performed early. The results of drug treatment for right-sided failure are very disappointing in congenital heart disease patients [3].
- LV dysfunctio; this is proportionate to the delay until total correction, the degree of cyanosis, and the period during which pulmonary flow is provided by a palliative shunt (Blalock, Potts, etc.). It rarely occurs if total correction is performed at an early age.
Tetralogy of Fallot and pulmonary agenesis
In this combination, there is no connection between the RV and PA – the RVOT is a dead end. This agenesis is combined with variable underdevelopment of the pulmonary artery, which is compensated by patent ductus arteriosus flow at birth. In extreme cases, the entire pulmonary flow is provided by a system of collaterals originating from the aorta without any link to the atretic pulmonary vascular tree (MAPCA, major aortopulmonary collateral arteries). R-to-L shunting through the VSD causes a complete mixture of venous and arterialised blood in the LV.
If possible according to patient's anatomy, surgical correction entails closing the VSD and connecting the RV to the PA using a valved conduit. In cases of MAPCA, unifocalisation is performed, whereby the collaterals are detached from the aorta and reimplanted in a conduit originating from the RV. The VSD is occluded. These children are managed in a similar way to those with univentricular physiology (see Ventricular Hypoplasia) [10].
Tetralogy of Fallot and absent pulmonary valve
This differs from the previous situation by a valveless connection between the RV and PA. Regurgitation is massive, the RV is dilated and the increase in PA pulsatility during foetal life causes aneurysmal dilation of the pulmonary vessels combined with bronchial compression and tracheomalacia. Correction involves reducing the size of the PA, closing the VSD, and creating a pulmonary valve (allograft). Management of such children involves maintaining contractility of the volume-overloaded RV and lowering PVR to limit pulmonary insufficiency (hyperventilation, low intrathoracic pressure). Any factors that may induce pulmonary vasoconstriction must be avoided.
Anaesthesia for palliation
If pulmonary flow is significantly reduced, the child cannot wait until the age of a total correction and must undergo temporary palliation in the form of a modified Blalock shunt. Instead of anastomosing the subclavian artery directly to the ipsilateral pulmonary artery, a 3-4 mm Goretex™ shunt is interposed between the two vessels on the opposite side to the aortic arch. This maintains normal perfusion in the arm and adjusts pulmonary flow based on the diameter of the tube.
Management is aimed at increasing SVR with systemic vasoconstrictors (norepinephrin, phenylephrine) to increase flow through the shunt and force VSD flow in a L-to-R direction. Beta stimulation is contraindicated as this would increase the dynamic component of the right infundibular stenosis. Surgery is performed by thoracotomy: it requires low-dose heparinisation (100 U/kg) and retraction of the lung, which reduces gas exchange. Once the shunt is open, systemic diastolic pressure falls (Figure 14.60).
Video: Systolo-diastolic flow in the left subclavian artery used for a Blalock shunt (short-axis view of the distal aortic arch).
Video: Systolo-diastolic continuous flow in a Blalock shunt between the subclavian artery and the pulmonary artery.
The palliative shunt imposes volume overload on the LV and inotropic support is often necessary. The required SpO2 is between 80% and 85%. A higher value indicates that pulmonary flow is excessive. However, even with acceptable SaO2, unilateral pulmonary oedema may develop on the side of the shunt. Continuation of positive-pressure ventilation for 24 hours postoperatively is recommended.
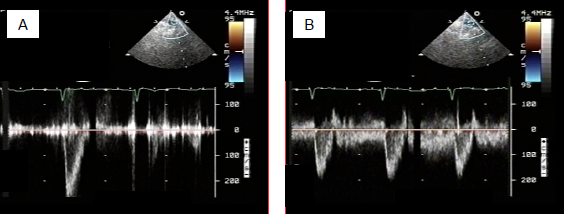
Figure 14.60: Flow during construction of a Blalock-Taussig shunt. A: flow in the subclavian artery before the shunt is constructed. The flow is solely systolic as in any systemic artery. B: after connection to the pulmonary artery, the flow becomes systolo-diastolic as in a LR shunt. Diastolic systemic arterial pressure consequently falls.
Anaesthesia for total correction
There are two objectives for managing a total correction – reducing the shunt and increasing pulmonary flow (see Table 14.5).
- Increasing systemic arterial resistance – this reduces the cyanotic R-to-L shunt, although at the cost of reducing systemic perfusion if vasoconstriction is intense. Moreover, arterial hypertension increases LV and RV afterload at a time when the RV is already under stress as it is subjected to systemic ejection pressure. SpO2 provides an effective and immediate means of monitoring the extent of shunting.
- Increasing pulmonary flow – arterial vasoconstriction (alpha-stimulants, phenylephrine) is indicated in cases of fixed obstruction. If the obstruction is dynamic, beta-blockade is also necessary to relax the RVOT musculature. Positive inotropic agents are contraindicated, as is atropine. Ventilation must be performed at a low intrathoracic pressure with hyperventilation to stimulate pulmonary flow.
- Maintaining normovolaemia – in the event of hypovolaemia, the shunt's R-to-L component increases as it does when systemic pressure falls. Moreover, these children exhibit polycythaemia and any dehydration further increases Ht, viscosity, and the risk of thrombosis. Crystalloid 10-15 mL/kg or albumin 5% is administered during induction [10].
A hypoxic episode (blue spell) may occur at any time. If not addressed in time, it may lead to cardiovascular collapse. Under general anaesthesia, the hypoxic crisis manifests as arterial desaturation, tachycardia, a reduction in arterial pressure, and loss of murmur on auscultation. Treatment must be immediate in the event of deep desaturation [10].
- Deepen anaesthesia (fentanyl) – avoid isoflurane, whose vasodilator effect is excessive.
- Ventilation at FiO2 1.0, ventilation at low intrathoracic Pmean.
- Phenylephrine 5-10 mcg/kg as an IV bolus, norepinephrin perfusion.
- Filling (15-30 mL/kg), raising the lower limbs.
- Bicarbonate (1-2 mEq/kg) if metabolic acidosis.
- If the pericardium is open, manual compression of the aorta.
- Esmolol (0.5 mg/kg, then 50-300 mcg/kg/min infusion).
Anaesthesia is based on a fentanyl-midazolam or fentanyl-sevoflurane combination. Induction may be performed with a mask (sevoflurane) or intravenously (ketamine) provided that there is no ventricular dysfunction or risk of blue spells. In haemodynamically unstable cases, the main agents should ideally be fentanyl (50-75 mcg/kg) or sufentanil (5.0-7.5 mcg/kg) with midazolam or etomidate as a back-up. Cyanosis is combined with coagulation defects and platelet inhibition. It potentiates systemic inflammatory response, limits haemodilution during CPB (risk of hypoxaemia) and forces to keep a high Ht for going off-bypass.
Several major problems are predominant postoperatively [10].
Several major problems are predominant postoperatively [10].
- RV systolic dysfunction – this is treated with dobutamine (5-10 mcg/kg/min) or epinephrin (0.01-0.1 mcg/kg/min) + milrinone (0.5-1.0 mcg/kg/min). Although useful in cases of right-sided stasis, nitroglycerin (2 mcg/kg/min) is not recommended for Fallot patients. Moreover, the hypertrophied RV in Fallot cases exhibits restrictive-type diastolic dysfunction in 30% of cases, which entails a significant elevation of CVP to maintain the ventricular output [2].
- Arrhythmias: atrial tachyarrhythmias, complete AV block requiring temporary pacing.
- Haemorrhage.
- Sequelae of surgery: ventriculotomy (dyskinesia of the RV free wall), pulmonary insufficiency (volume overload of the RV), residual VSD (ditto).
If palliation has previously been performed by means of a systemic-pulmonary shunt or if large aortopulmonary collaterals are present, pulmonary perfusion is dependent on satisfactory systemic pressure. Any arterial vasodilation lowers the shunt flow, thereby also reducing pulmonary flow – cyanosis increases. These children therefore also benefit from increased SVR. Diastolic arterial pressure is chronically lowered if the shunt is flowing satisfactorily, since pressure in the pulmonary vascular tree is constantly diverting blood flow. Since the LV must ensure systemic and pulmonary perfusion, it functions in a situation of volume overload, bordering on failure.
| Tetralogy of Fallot) |
|
Characteristics:
- Subaortic VSD (membranous septum); - Aorta overriding the VSD - Pulmonary stenosis (RVOT, pulmonary valve, PA trunk) - RVH (RV connected to systemic pressure) Cyanotic R → L shunt, reduced pulmonary blood flow (Qp ↓, Qp/Qs < 1.0) In order to reduce the shunt, increase SVR (alpha stimulation) and reduce PVR (hyperventilation). If dynamic component (RVOT obstruction): beta-blocker SaO2 proportionate to Qp/Qs Anaesthesia recommendations: - Sevoflurane or fentanyl/midazolam GA (or etomidate for induction) - Ventilation: hypocapnia, FiO2 has no significant effect on SpO2 - Increase SVR to ↑ SpO2 - Prevent systemic vasodilation, vasoconstrictor infusion if required - Maintain afterload: hypovolaemia increases cyanotic R → L shunting If pulmonary flow is provided by a palliative shunt (e.g. Blalock-Taussig) or collaterals from Ao to PA (fixed flow): - Qp proportionate to systemic arterial pressure - Keep SVR ↑ - Hypovolaemia/hypotension: Qp ↓, cyanosis ↑ |
© BETTEX D, BOEGLI Y, CHASSOT PG, June 2008, last update February 2020
References
- AL HABIB HF, JACOBS JP, MAVROUDIS C, et al. Contemporary patterns of management of tetralogy of Fallot: data from the Society of Thoracic Surgeons database. Ann Thorac Surg 2010; 90:813-20
- CHATURVEDI RR, SHORE DF, LINCOLN C, et al. Acute right ventricular restrictive physiology after repair of tetralogy of Fallot : association with myocardial injury and oxidative stress. Ciculation 1999; 100:1540-7
- DAVLOUROS PA, NIWA K, WEBB G, GATZOULIS MA. The right ventricle in congenital heart disease. Heart 2006; 92(Suppl 1): i27-i38
- DIDONATO RM, JONAS RA, LANG P, ROWE JJ, CASTANEDA AR. Neonatal repair of tetralogy of Fallot with and without pulmonary atresia. J Thorac Cardiovasc Surg 1991; 101:126-37
- ERIKSSEN G, LIESTOL K, SEEM E, et al. Achievements in congenital heart defect surgery: a prospective, 40-year study of 7'038 patients. Circulation 2015; 131:337-46
- HONJO O, Van ARSDELL GS. Cardiovascular procedures : surgical considerations. In : BISSONNETTE B, edit. Pediatric anesthesia. Basic principles, State of the art, Future. Shelton (CO): People’s Medical Publishing House (USA), 2011, 1589-608
- JIMENEZ M, ESPILl G, THAMBO JB, et al: Outcome of operated Fallot's tetralogy. Arch Mal Coeur Vaiss 2002; 95:1104-11
- KHAMBADKONE S. Percutaneous pulmonary valve implantation. Ann Pediatr Cardiol 2012; 5:53-60
- KLOESEL B, DINARDO JA, BODY SC. Cardiac embryology and molecular mechanisms of congenital heart disease – A primer for anesthesiologists. Anesth Analg 2016; 123:551-69
- NASR VG, DINARDO JA. The pediatric cardiac anesthesia handbook. Oxford: Wiley-Blackwell, 2017; 131-46
- PIGULA FA, KHALIL PN, MAYER JE, et al. Repair of tetralogy of Fallot in neonates and young infants. Circulation 1999; 100(Suppl II):157-61
- SILVERSIDES CK, KIESS M, BEAUCHESNE L, et al. Canadian Cardiovascular Society 2009 Consensus Conference on the management of adults with congenital heart disease: Outflow tract obstruction, coarctation of the aorta, tetralogy of Fallot, Ebstein anomaly and Marfan’s syndrome. Can J Cardiol 2010; 26:e80-e97
- SYFRIDIS PG, KIRVASSILIS GV, PAPAGIANNIS JK, et al. Preservation of right ventricular structure and function following transatrial transpulmonary repair of tetralogy of Fallot. Eur J Cardiothorac Surg 2013; 43:336-42
- VAN PRAAGH R. Tetralogy of Fallot: underdevelopment of the pulmonary infundibulum and its sequelae. Am J Cardiol 1970; 47:1279
- VILLAFAÑE J, FEINSTEIN JA, JENKINS KJ, et al. Hot topics in tetralogy of Fallot. J Am Coll Cardiol 2013; 62:2155-66
- WARNES CA. Adult congenital heart disease. Importance of the right ventricle. J Am Coll Cardiol 2009; 54:1903-10
- WEINTRAUB R, SHIOTA T, ELKADI T, et al. Transesophageal echocardiography in infants and children with congenital heart disease. Circulation 1992; 86:711-22