A new concept emerged about fifteen years ago, based on the predominant role of cellular elements [3,5]. It provides a better understanding of how coagulation remains localised to the injured vessel without spreading instantaneously throughout the vascular tree. It is based on the triggering role of tissue factor (TF) exposed by an endothelial injury and that of the TF/VIIa complex anchored to a parietal cell normally sheltered from blood stream by the endothelium [4]. It involves two cell types:
- The tissue cell carrying TF (fibrocytes, myocytes, lipocyte, mononuclear, macrophage, collagen);
- The circulating thrombocyte, which provides a negatively charged phospholipid surface for anchoring coagulation factors.
The cellular pathway consists of three partially overlapping phases: initiation, amplification and propagation.
Initiation
The contact between two normally separated cell types, subendothelial cells and thrombocytes, is the key to initiating coagulation. TF-carrying cells come into contact with blood elements upon endothelial injury (trauma, unstable plaque). TF, a 47,000 Da transmembrane glycoprotein, activates factor VII (FVIIa), which immediately binds to it. This TF/VIIa complex in turn activates factors IX (FIXa) and X (FXa). The latter associates with its cofactor, factor Va, which self activated; calcium (Ca2+ ) is required for this sequence. The Va/Xa complex forms the prothrombinase complex, which is attached to the TF-bearing parietal cell. This complex triggers the conversion of prothrombin (factor II) to thrombin (factor IIa) (Figure 8.2). This phase lasts 5-7 minutes.
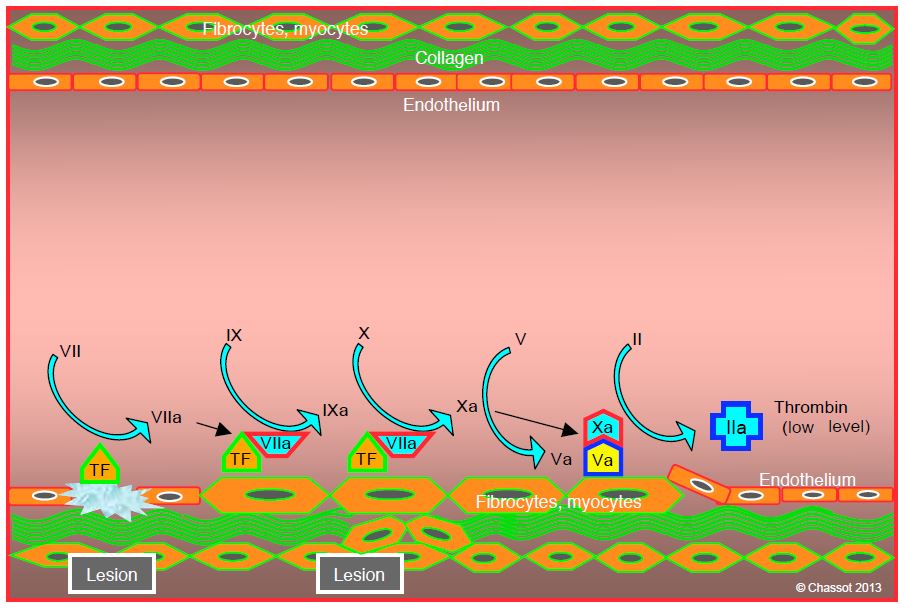
Figure 8.2: Initiation phase. Tissue factor (TF) carrier cells are brought into contact with blood during an endothelial injury (trauma, unstable plaque rupture). TF activates factor VII (fVIIa), which immediately sticks to it. This TF/VIIa complex in turn activates factors IX (fIXa) and X (fXa). The latter associates with its cofactor, factor Va, which it has itself activated. The combination of Va/Xa forms the prothrombinase complex, which is attached to the parietal cell. This complex triggers the transformation of prothrombin (factor II) into thrombin (factor IIa), which level is still minimal during this phase.
At this stage, thrombin appears in minimal quantities (picomolar concentration) and does not lead to clot formation, because thrombocytes, factor VIII and von Willebrand factor are not yet activated. They circulate in blood in soluble and inactive form. The sequence described so far remains localised to the endothelial lesion. Factor Xa molecules that escape into the bloodstream are rapidly inhibited by antithrombin (AT III), whereas factor IXa is blocked much more slowly by AT III. The proliferation of the TF/VIIa complex is limited by its circulating inhibitor (TFPI, tissue factor pathway inhibitor) (see Regulatory systems).
Amplification
The amplification phase takes place on the surface of platelets. It starts when the vascular injury is sufficient for platelets to be activated and for von Willebrand factor to cross the endothelial barrier and adhere to TF-bearing cells. The small amount of thrombin formed during the initiation phase has several functions (Figure 8.3) [3,5].
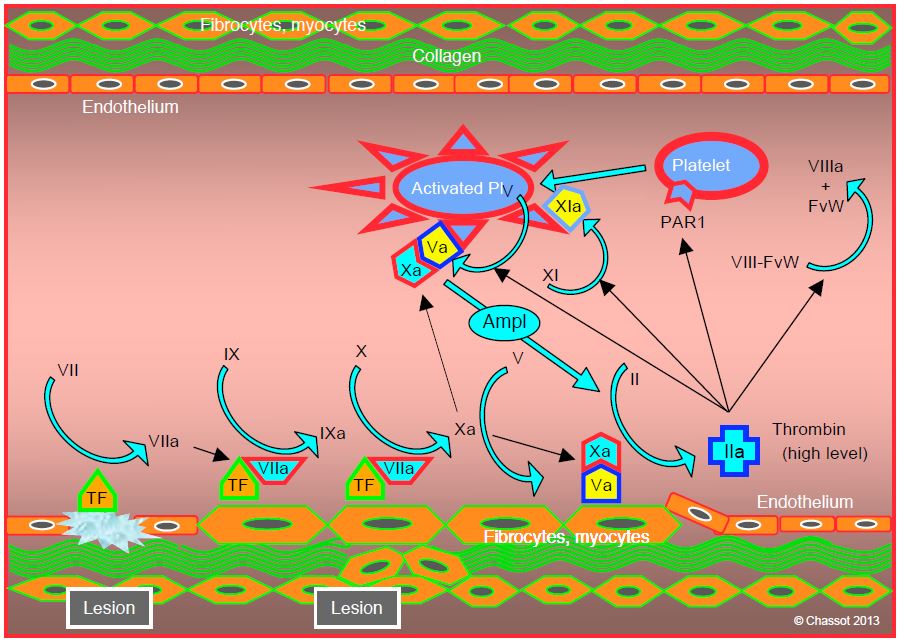
Figure 8.3: Amplification phase. The small amount of thrombin formed during the initiation phase has several functions: 1) activation of platelets by stimulation of their thrombin receptors (PAR1); 2) activation of thrombocyte factor V; fVa binds to locally produced fXa to form a prothrombinase complex (Va/Xa); 3) activation of platelet factor XI (XIa); and 4) dissociation of the circulating factor VIII/von Willebrand factor complex (VIII/vWF). The presence of the prothrombinase complex (Va/Xa) on the platelet surface creates an amplifying system (Ampl), as it converts prothrombin (fII) to thrombin (fIIa), which in turn causes platelet activation. TF: tissue factor.
- Activation of platelets by stimulation of their receptors for thrombin (PAR-1, protease-activated receptor-1); they change shape and develop spicules.
- Activation of factor V in thrombocytes, with the activated form (Va) appearing on their surface; FVa binds to locally produced FXa to form a prothrombinase complex (Va/Xa).
- Activation of platelet factor XI (XIa).
- Dissociation of the circulating factor VIII/von Willebrand factor (VIII/VWF) complex; vWF, also present in subendothelial tissues, allows platelet adhesion to the site of vascular injury via its GP Ib/V/IX receptor. Factor VIIIa remains bound to the platelet surface.
The presence of the prothrombinase complex (Va/Xa) on the surface of platelets creates an amplifying system, as it converts prothrombin to thrombin, which in turn causes platelet activation. This positive feedback exponentially increases the number of activated platelets at the site of vascular injury. The presence of calcium (Ca2+ ) is required for the prothrombinase complex (Va/Xa) to convert prothrombin to thrombin. The central position of factor Xa as a trigger for amplification explains the impact on coagulation of substances that inhibit it directly, such as rivaroxaban or apixaban.
Propagation
The propagation phase is characterised by fibrin formation and clot formation at the endothelial lesion. It takes place on the surface of the platelets (Figure 8.4) [3,5].
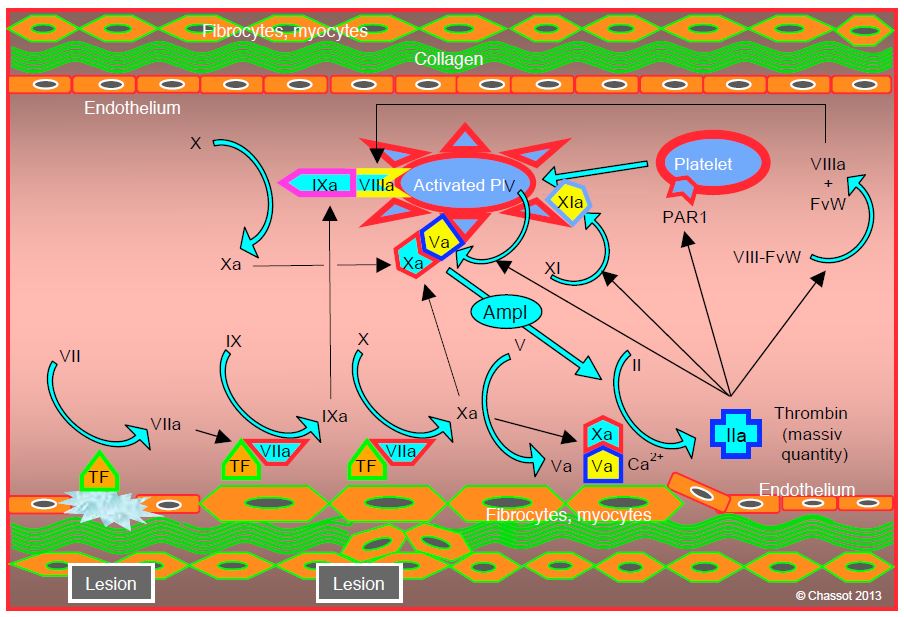
Figure 8.4: Propagation phase, characterised by fibrin formation and clot formation at the endothelial lesion. It takes place on the surface of the platelets: 1) binding of factor IXa produced during the initiation phase, with factor VIIIa dissociated from von Willebrand factor (IXa/VIIIa complex or tenase complex); 2) production of more factor Xa by the IXa/VIIIa complex; 3) binding of factor Xa with platelet factor Va and formation of prothrombinase Va/Xa complex in the presence of Ca2+ ; 4) factor XIa promotes the formation of factor IXa on the surface of thrombocytes, which will increase the appearance of factor Xa; the latter has a central role in coagulation. The result of this activity is a surge in thrombin production (thrombin burst), powerful enough to trigger the massive transformation of fibrinogen into fibrin.
- Binding of factor IXa produced during the initiation phase with factor VIIIa dissociated from von Willebrand factor; this IXa/VIIIa complex (tenase complex) is located on the surface of platelets.
- Production of more factor Xa by the IXa/VIIIa complex.
- Binding of factor Xa with platelet factor Va and formation of Va/Xa prothrombinase complex.
- The role of factor XIa is to promote the formation of factor IXa on the surface of thrombocytes, which will increase the appearance of factor Xa.
There are therefore several positive feedback loops. The result of this activity is a thrombin burst, powerful enough to trigger the massive conversion of fibrinogen to fibrin. The platelet is central to this phase, as it alone provides the adequate surface area and positioning required to forge the tenase and prothrombinase complexes. The attachment of activated and aggregated platelets to the endothelial lesion occurs in parallel with the stimulation of the coagulation chain. The essential condition for this system to function is the presence of sufficient fibrinogen and platelets.
Mass-produced thrombin cleaves fibrinogen molecules into fibrin monomers that spontaneously polymerise into an uncross-linked fibrin gel. Each thrombin molecule can activate 1,000 fibrinogen molecules. The degree of final stability of the thrombocyte-fibrin combination, which determines the quality of haemostasis and the end of bleeding, depends on the local conditions: pH, temperature, calcium concentration (Ca2+ ), quantity of thrombin, platelets, fibrinogen and factor XIII [6]. Factor XIII activated by thrombin and Ca2+ (XIIIa) provides the bulk of this stability (Figure 8.5). It creates bridges between the fibrin elements (cross-linked fibrin) and makes them rigid [2]. A second stabilising factor is the thrombin-activated fibrinolysis inhibitor (TAFIa), which reduces the attachment points for plasmin. Both factors protect thrombus from fibrinolysis.
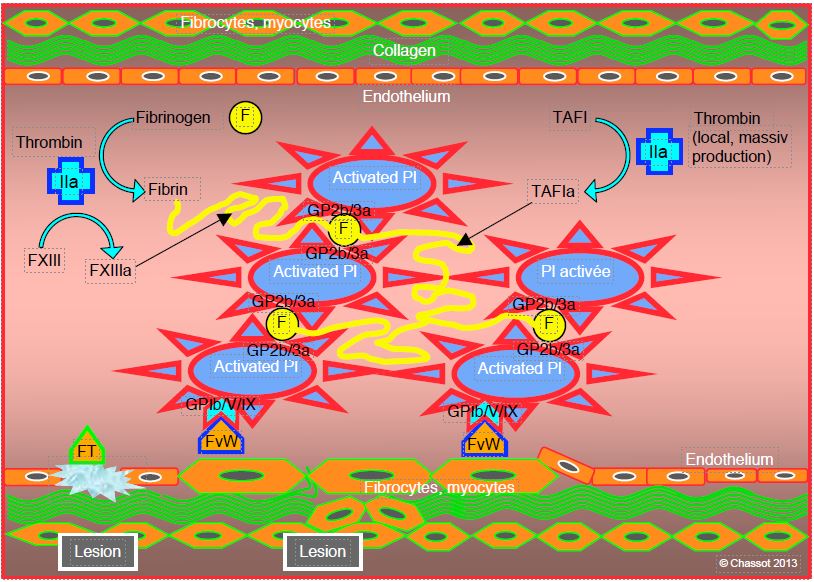
Figure 8.5: Fibrin formation. Activated platelets, bound to the endothelial lesion by von Willebrand factor (vWF) bound to the GP Ib/V/IX receptor, provide mechanical support for the coagulation chain. They are aggregated by the link between their GP IIb/IIIa receptors and fibrinogen (F). Mass-produced thrombin (IIa) cleaves fibrinogen molecules into fibrin monomers which spontaneously polymerise into an uncross-linked fibrin gel. A second stabilising factor is the thrombin-activated fibrinolysis inhibitor (TAFIa), which reduces the attachment points for plasmin.
Platelets
Platelets and fibrinogen molecules circulate smoothly in the bloodstream as long as the endothelium is intact. A break in the continuity of the endothelium, whether due to a vascular breach or unstable atheromatous plaque, brings elements normally hidden in the vessel wall into contact with blood stream: collagen, von Willebrand factor (vWF), macrophages, lipids, etc. This triggers platelet activation, which proceeds in three phases (Figure 8.6) [1].
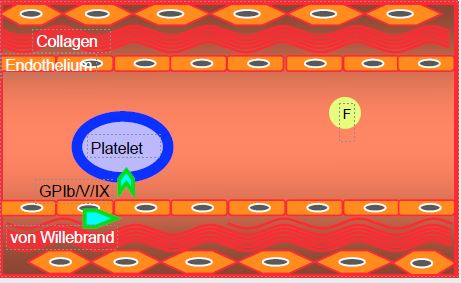
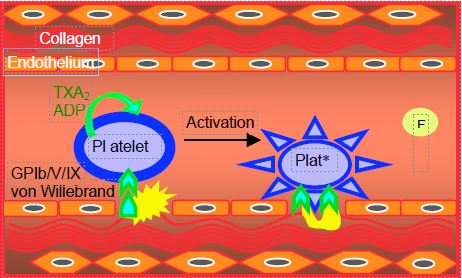
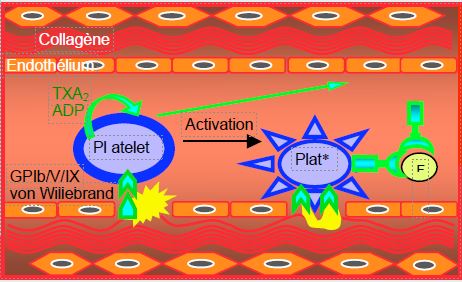
Figure 8.6: Platelet activation and platelet thrombus (white thrombus) formation in endothelial injury exposing collagen normally covered by endothelium: inactivated platelets and factors (8.6 A), initiation phase (8.6 B) and extension phase (8.6 C). Antiplatelet agents (aspirin, clopidogrel, etc) act by blocking the activation (or initiation) phase of platelets. Plaq*: activated platelet.
- Initiation phase, during which platelets adhere to the lesion; von Willebrand factor is one of the main elements that bind platelets to the vascular lesion;
- Extension phase, characterised by recruitment and aggregation of other platelets; a white thrombus is formed (platelets bound together by fibrinogen molecules);
- Perpetuation phase, marked by stabilisation of thrombus through fibrin (red thrombus).
When endothelial injury occurs, platelets come into contact with collagen and von Willebrand factor. The encounter between subendothelial structures and circulating thrombocytes specifically activates the GP Ib/V/IX receptor and causes platelet degranulation, which releases thromboxane A2 (TXA2 ), adenosine diphosphate (ADP) and thrombin. These substances in turn stimulate the corresponding receptors on each platelet and on neighbouring thrombocytes, thereby amplifying the reaction by recruitment of new elements [7]. Activated platelets change their shape and develop typical spicules.
Stimulation of platelet receptors by TXA2 , ADP and thrombin transmits an intracellular signal that leads to an increase in sarcoplasmic ionised Ca2+ and a decrease in cyclic AMP (cAMP) production; this activates the several thousand GP IIb/IIIa complexes located on the surface of each platelet. Once activated, these complexes bind to circulating fibrinogen. This binding forms bridges between several platelets and becomes the glue that binds thrombocytes together. The chain thus formed initiates the platelet plug, the skeleton of the future clot.
When stimulated, platelets release coagulation activating agents (TXA2 , ADP, serotonin, thrombin), some of which are also local vasoconstrictors (TXA2 , serotonin), while the endothelium secretes substances that inhibit platelet activity and have a vasodilator effect: NO• (decrease in intracellular ionised Ca2+ ), prostacyclin PGI2 (increase in cAMP, modulation of the response to TXA2 ) and ecto-ADPase (suppression of the platelet recruitment phase).
Termination
Clotting must remain limited to the area of the lesion and not spread to the entire vascular tree. Several systems prevent this spread (see Regulatory systems) (Figure 8.7).
- Thrombin is rapidly inactivated by antithrombin III (ATIII) and by thrombomodulin (TM);
- The TM-thrombin complex activates protein C which blocks factors Va and VIIIa;
- The TF/VIIa complex is limited by its circulating inhibitor (TFPI, tissue factor pathway inhibitor) which rapidly inhibits any circulating complex outside the injured area.
In the long term, fibrinolysis brings the progressive degradation of fibrin clusters and resorption of thrombus.
Virchow Triad
The Virchow triad (Rudolf Virchow, 1821-1902) summarises three elements involved in thrombosis: vessel wall, blood and blood flow. Arterial and venous thrombosis do not have the same genesis and are rarely simultaneous [8].
- Arterial thrombosis: is associated with a rupture of atheromatous plaque which brings elements normally sheltered by endothelium into contact with blood: tissue factor, collagen, macrophage, lipocytes, lipids. The mural thrombus, initially formed by platelets, creates a stenosis that causes shear stress and turbulence, potentially generating more thrombus.
- Venous thrombosis: typically associated with hypercoagulability, is related to procoagulant activity of endothelium following inflammatory stimulation; slowing of blood flow is a key element.
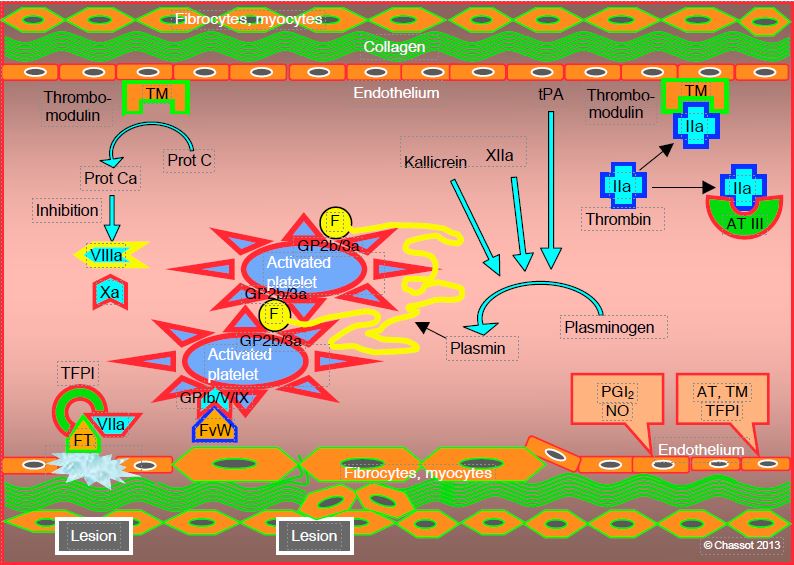
Figure 8.7: Limitation. Several systems prevent the diffusion of coagulation: 1) endothelial thrombomodulin (TM) inhibits circulating thrombin; 2) the TM-thrombin complex activates protein C (Prot Ca) which blocks factors Va and VIIIa; 3) antithrombin III (AT III) rapidly inactivates thrombin and more slowly factor IXa; 4) proliferation of the TF/VIIa complex is limited by its circulating inhibitor (TFPI, tissue factor pathway inhibitor); 5) fibrinolysis ensures the progressive degradation of the fibrin clusters and the resorption of the thrombus; it is ensured by plasmin derived from plasminogen under the action of kallikrein, factor XIIa and the endothelial activator (tPA, tissue plasmin activator). PG: prostaglandin.
| Cellular pathway of coagulation |
|
Coagulation takes place in 3 phases (initiation, amplification and propagation) which develop on the surface of two types of cells:
- Extravascular cells normally isolated by endothelium (fibrocytes, macrophages)
- Activated circulating platelets
1 - Endothelial damage brings tissue factor (TF) carrier cells into contact with blood. The coagulation chain is activated (initiation):
- TF/FVIIa complex formation (extrinsic pathway)
- Activation of factor X to F Xa
- Formation of Xa/Va complex (prothrombinase complex)
- Transformation of prothrombin (factor II) to thrombin (F IIa) (minimal amount)
2 - The minimal amount of thrombin formed has several effects (amplification):
- Platelet activation
- Release of F VIIIa (from complex with von Willebrand factor)
- Activation of platelet factor V to F Va and F IX to F IXa
- Formation of Xa/Va complex (prothrombinase complex)
- Positive feedback on the transformation of F II to F IIa (thrombin) (large amount)
3 - Thrombin is produced en masse; it converts fibrinogen into fibrin, which forms a clot with platelets (propagation):
- IXa/VIIIa complex formation (intrinsic pathway)
- Formation of more Xa/Va complex (prothrombinase complex)
- Positive feedback with massive F Xa and thrombin formation
- Cleavage of fibrinogen into fibrin, which polymerises and forms a gel enclosing platelets
The aggregation of platelets also proceeds in 3 phases:
- Adhesion to the vascular lesion by GP Ib/V/IX receptors
- Platelet recruitment and aggregation by binding of GB IIb/IIIa receptor to fibrinogen molecules (white thrombus)
- Stabilisation by fibrin (red thrombus)
|
© CHASSOT PG, MARCUCCI Carlo, last update November 2019.
References
- ANGIOLILLO DJ, UENO M, GOTO S. Basic principles of platelet biology and clinical implications. Circ J 2010; 74:597-607
- BAGOLY Z, KONCZ Z, MUSZBEK L. Factor XIII, clot structure, thrombosis. Thromb Res 2012; 129:382-7
- HOFFMAN M. Remodeling the blood coagulation cascade. J Thromb Thrombolysis 2003; 16:17-20
- HOFFMAN M. A cell-based model of coagulation and the role of factor VIIa. Blood Reviews 2003; 17 (Suppl 1):S1-S5
- HOFFMAN M, MONROE DM. A cell-based model of hemostasis. Thromb Haemost 2001; 85:958-65
- INNERHOFER P, KIENAST J. Principles of perioperative coagulopathy. Best Pract Res Clin Anaesthesiol 2010; 24:1-14
- Sakhuja R, YEH RW, BHATT DL. Antiplatelet agents in acute coronary syndromes. Curr Probl Cardiol 2010; 35:123-70
- WOLBERG AS, ALEMAN MM, LEIDERMAN K, MACHLUS KR. Procoagulant activity in hemostasis and thrombosis: Virchow's triad revisited. Anesth Analg 2012; 114:275-85