Clinical data
Pulmonary arterial hypertension (PAH) is defined in infants aged 3 months and above as mean resting PAP > 25 mmHg and/or pulmonary vascular resistance (PVR) > 3 Wood Units or 300 dynes•cm•s-5 (see Chapter 12 Pulmonary Hypertension) [10,20]. In paediatrics, the two most common aetiologies are congenital heart diseases (50-70% of cases) and primary idiopathic PAH (25% of cases) [25,26]. PAH in paediatric congenital patients is included in group 1 of the pulmonary hypertension (PHT) classification [20,43].
Pulmonary arterial hypertension (PAH) is defined in infants aged 3 months and above as mean resting PAP > 25 mmHg and/or pulmonary vascular resistance (PVR) > 3 Wood Units or 300 dynes•cm•s-5 (see Chapter 12 Pulmonary Hypertension) [10,20]. In paediatrics, the two most common aetiologies are congenital heart diseases (50-70% of cases) and primary idiopathic PAH (25% of cases) [25,26]. PAH in paediatric congenital patients is included in group 1 of the pulmonary hypertension (PHT) classification [20,43].
- 1 - Pulmonary arterial hypertension (PAH): congenital heart diseases, primary idiopathic PAH, drug/infection/age-related PAH.
- 1’ - Veno-occlusive pulmonary disease.
- 1’’ - Persistent PAH in neonates (2: 1,000 babies).
- 2 - Post-capillary pulmonary hypertension (PHT) (LVEDP > 18 mmHg, PAOP > 15 mmHg, but PVR < 3 Wood Units and transpulmonary gradient < 12 mmHg): systolic failure or diastolic insufficiency of the LV, mitral valvular disease.
- 3 - PAH combined with alveolar hypoxia: COPD, emphysema, ARDS, sleep apnoea syndrome, high altitude hypoxia.
- 4 - PAH related to chronic pulmonary thromboembolic disease.
- 5 - Unexplained multi-factorial PAH.
The annual incidence of paediatric PAH is 64 cases per million children [48]. The most common cause of PAH in congenital heart diseases is a non-restrictive ventricular or arterial L-to-R shunt: VSD, atrioventricular canal defects, truncus arteriosus, surgical aortopulmonary shunts, transposition of the great arteries + VSD [36]. Pulmonary hypertension exhibits a number of haemodynamic characteristics [7,11,17,29].
- Pulmonary blood flow is reduced and fixed. Any compliance with variations in blood flow and circulating volume is lost. It is impossible to increase Qp if there is an increased demand for O2: children experience hypoxaemia at the slightest effort.
- The more hypertrophied is the RV, the more it behaves like the LV. Its Frank-Starling curve is steeper and its blood flow becomes dependent on its preload. In such cases, hypovolaemia causes a fall in pulmonary blood flow.
- In L-to-R shunts, reduced systemic pressure due to hypovolaemia or systemic vasodilation exacerbates the shunt's R-to-L component triggering cyanosis. Systemic hypotension lowers coronary perfusion pressure, increases the interventricular septum’s shift to the left, and threatens subendocardial perfusion of the RV.
- During diastole, pressure in the hypertrophied and overloaded RV is higher than the pressure in the LV. The interventricular septum bulges into the LV (Bernheim effect) reducing its diastolic filling (see Figure 12.8A). It is usually possible to replace the septum in its physiological position by increasing left-side afterload through systemic vasoconstriction.
- Peripheral pulmonary arteriole reactivity is preserved despite the thickening of vessel walls – hypercapnia, acidosis and hypoxaemia further increase PVR while hyperventilation reduces it [7,8]. Anaesthetists must therefore avoid any procedures that might increase PVR [11]:
- Hypoventilation: hypercapnia, hypoxaemia, atelectasis;
- High intrathoracic pressure;
- Acidosis;
- Hypothermia;
- Anaemia (Hb < 100 g/L);
- Sympathetic stimulation, stress, pain.
Children aged under one year with high pulmonary blood flow linked to an L-to-R shunt are at very high risk of paroxysmal pulmonary hypertensive crises in the perioperative and postoperative period, in some cases requiring ECMO (PAH treatment – Table 14.7). A number of criteria may be applied to ascertain PAH-related clinical risk. This risk is increased in the following circumstances [6,26].
- Clinical signs of RV failure;
- Fainting;
- Growth retardation;
- Functional class 3/4;
- Raised NT-proBNP;
- Echocardiography: RV dilation, bulging of the interventricular septum into the LV, reduction of tricuspid annular plane systolic excursion (TAPSE), deterioration of RV functional indices on tissue Doppler [28,44];
- Haemodynamics: mPAP/MAP ratio > 0.7, RAP > 10 mmHg, PVR > 8 Wood Units, CO < 2.5 L/min/m2.
An increase in pulmonary blood flow and compression of the airways by dilated and hypertrophied vessels lead to reduced lung compliance, increased airway resistance, and increased work of breathing. The severity of these consequences determines operability and risks associated with the postoperative period. If surgical correction is performed early, PAH is gradually corrected. However, if the mPAP/SAP ratio is already > 0.7 (PVR/SVR > 0.3), it is not possible to operate [1]. Operability is also determined by a pulmonary vasodilation test with NO or prostaglandins. The test is considered as positive if mPAP falls by at least 20% without lowering cardiac output, provided that there is no massive shunt at ventricular or ductal level [1,26]. In this case, a post-operative PAH relapse is likely.
| Pulmonary arterial hypertension (PAH) |
|
Definition of PAH: PAPmean > 25 mmHg at rest. Main aetiologies in paediatrics: non-restrictive L-to-R shunt, idiopathic PAH, persistent PAH in neonates.
Haemodynamic characteristics - Low and fixed pulmonary blood flow - Pulmonary vascular reactivity preserved - RVH: RV outuput is dependent on its preload - Systemic hypotension: exacerbation of the R-to-L shunt component + cyanosis, risk of ischaemia and loss of left-side assistance for the RV Triggers of PAH crises - Hypoventilation (hypoxaemia, hypercapnia, atelectasis) - High intrathoracic pressure - Acidosis - Hypothermia - Stress, sympathetic stimulation, pain |
Chronic treatment of pulmonary hypertension in paediatrics
Basic chronic treatment involves a range of medications that reduce pulmonary vascular resistance combined with a non-specific general treatment [1,26,31].
- Calcium channel blockers (amlodipine 2.5-7.5 mg/d, nifedipine 2-3 mg/kg/d, diltiazem
- 3-5 mg/kg/d). These are only indicated if the NO reactivity test is positive. They are the first choice for idiopathic PAH. They are contraindicated for infants aged under 12 months due to their negative inotropic effect and are not recommended for congenital heart diseases since they cause major systemic vasodilation, which may increase the R-to-L component of a shunt.
- Endothelin receptor antagonists: bosentan (1-2 mg/kg bid), ambrisentan (0.05-0.1 mg/kg 1 x/d, max 10 mg); 2 years and above [25,41]; sitaxentan has been withdrawn from the market.
- Phosphodiesterase-5 inhibitors: sildenafil (0.5-1 mg/kg tid), tadalafil (0.5-1 mg/kg/d); 12 months and above.
- Prostaglandins: inhaled (iloprost, treprostinil), oral (iloprost, treprostinil), sub-cutaneous (treprostinil), or intravenous (epoprostenol, treprostinil). Continuous intravenous prostaglandins are the most powerful pulmonary vasodilators. Dosages: epoprostenol
- 25-50 ng/kg/min infusion; treprostinil 25-50 ng/kg/min IV or subcutaneous; iloprost
- 6-9 inhalations (2.5-5.0 mcg/dose) per day.
- New treatments being investigated – no clinical data for children.
- Stimulators of vasodilator cGMP (riociguat);
- Growth factor antagonists (imatinib), inhibiting pulmonary vessel remodelling;
- Prostacyclin receptor agonists (selexipag).
- Non-specific treatment: diuretics (furosemide, thiazide, spironolactone), anticoagulation (vitamin K antagonists for INR 1.5-2.0), digoxin (5 mcg/kg bid < 10 years), O2 (if PaO2 < 60 mmHg or SaO2 < 92%).
- Atrial septostomy – the atrial septum is perforated by catheterisation or surgery to relieve the RA by draining its overload into the LA and increase the LV blood flow by means of an R-to-L shunt. However, the drawback of this procedure is arterial desaturation, since the resulting shunt is cyanotic (approximately 10% reduction in SaO2) [2].
- Potts shunt between the root of the descending aorta and the root of the left PA. This is indicated if PAP is suprasystemic. This operation reduces RV afterload, increases LV afterload, and repositions the interventricular septum to the right. It has the benefit of maintaining a supply of normally oxygenated blood to the coronary and carotid arteries, since arterial desaturation only affects the viscera and lower limbs. The mortality rate for this operation is 25% [5].
- Implantation of a stent in the ductus arteriosus if it is still patent. The factors to be considered are the same as for the Potts shunt.
- Aortic banding – moderate stenosis of the aortic root increases LV afterload and pushes the interventricular septum into the RV due to increased left ventricular pressure. By bolstering the RV, this reduces its dilation and increases its systolic performance.
- Lung transplant, generally bilateral. Mean survival is 49% at 5 years [47].
Acute treatment of pulmonary hypertension
Anaesthetists must take all possible steps to reduce pulmonary pressure, both perioperatively and in the operating theatre. These are listed in Table 14.7 [11]. It is essential for patients to be deeply anaesthetised (stress-free anaesthesia, high-dose fentanyl, sufentanil or remifentanil) and curarised. They must remain normothermic. The first stage in acute treatment of PAH involves ventilation, since respiratory alkalosis, gas inhalation and substance nebulisation are the only techniques capable of lowering pulmonary arterial pressure without causing systemic hypotension. Moreover, inhalation offers the benefit of not impeding hypoxic pulmonary vasoconstriction in non-ventilated areas, since substances are only distributed to the ventilated alveoli. Consequently, hypoxaemia is not exacerbated [21].
- Hyperventilation: hypocapnia (PaCO2 30-35 mm Hg) and alkalosis (pH 7.5) have a pulmonary vasodilator effect without affecting the systemic vascular bed. FiO2 must be high, ventilation pressure must be low and the tidal volume must be similar to the FRC (6-8 mL/kg.). pH rather than pCO2 regulates the pulmonary vascular tone. PVR reactivity to acidosis and alkalosis is increased in cases of hypoxia and pulmonary hypertension [7]. Ideally, the ventilator should be set to ensure a minimal mean ventilation pressure (6-10 mmHg) for PaCO2 of 30-35 mmHg.
- Administering O2 through a mask outside the operating theatre is beneficial, especially in severe cases of erythrocytosis (Ht > 55%).

This is followed by drug treatment. Some substances can be nebulised into the respiratory circuit and therefore have no systemic impact [20].
- NO•: 10 - 40 ppm in the ventilator's inspiratory circuit. The NO is immediately inactivated by its bond with haemoglobin, but it impairs platelet function. In cases of PAH or situations where PAP needs to be kept low (e.g. Fontan), NO• must be administered on induction and/or resumption of ventilation after CPB, before any hypertensive crises occur [12]. If PVR does not fall within 30 minutes of administering NO•, this indicates that the substance is ineffective and treatment must be discontinued [4]. Withdrawal must be performed gradually and may be difficult due to a rebound effect in terms of PAH. This may be mitigated by administering sildenafil, prostacyclin or milrinone.
- Prostacyclins: iloprost as a nasal spray (Ilomedin®, 2.5-5.0 mcg in 15 min 6-8 x/d), or in continuous nebulisation (0.2-0.3 mL/min of a 10-20 mcg/mL solution); treprostinil
- (2.5 ng/kg/min); epoprostenol 50 ng/kg/min for 15 minutes. In acute treatment, prostacyclins have an additive effect with NO•. They also reduce platelet aggregability.
- Phosphodiesterase-3 inhibitors – when nebulised rather than infused, milrinone exhibits less of a systemic hypotensive effect and greater pulmonary efficacy. Nebulisation of a 1 mg/mL solution at 0.2-0.3 mL/min for 10-20 minutes, i.e. 50-80 mcg/kg in 10 min. It is less expensive than NO and prostaglandins, and does not cause any rebound effects when discontinued. Iloprost and milrinone are also easier to use than NO•.
- Prostaglandins (PGE1): by stimulating adenylyl cyclase, these increase cAMP levels and prompt vasodilation. When used in chronic and acute treatment, they have an additive effect with NO• [39, 42].
- Epoprostenol (Flolan®) as an infusion (2-5 ng/kg/min) by central line; very short half-life (4-6 min);
- Iloprost (Ilomedin®) IV (0.5-3.0 ng/kg/min); duration of action: 60 minutes;
- Treprostinil (Remodulin®) subcutaneous or IV (1.25-2.5 ng/kg/min); 3-hour half-life.
- Phosphodiesterase-5 inhibitors: sildenafil (Revatio®, 3 x 10 mg/d IV < 10 kg, 20 mg 3 x/d > 20 kg), particularly effective when combined with L-arginine. It is used concurrently with NO• and prostaglandins. It is useful for preventing a rebound effect on discontinuation of NO [22,32].
- Magnesium (gluconate, sulphate): 20 mg/kg/hour.
- Arginine: through its transformation into citrulline, arginine chloride (15 mg/kg/min) provides the substrate required for producing NO•. It is probably indicated after CPB as this lowers arginine levels.
- Citrulline: this NO• precursor is capable of reducing PAH in cases of paediatric congenital heart disease (9 mg/kg/hour infusion). Only limited data are currently available.
- Calcium channel blockers may be useful for treating children with idiopathic PAH who exhibit positive vasodilatory response to an NO test (12-40% of cases). Contraindications: age < 12 months, risk of R-to-L shunt by systemic vasodilation [25].
- In general, pulmonary vasodilators are not indicated for patients with post-capillary PHT linked to left heart diseases (pulmonary venous obstruction, cor triatriatum, mitral stenosis, raised LAP due to LV outflow tract obstruction), as increased pulmonary blood flow would increase stasis in the LA and pulmonary veins. Only PDE5 inhibitors are capable of a beneficial effect on post-capillary PHT provided that the transpulmonary gradient (mPAP - PAOP) is > 12 mmHg [23].
- To reduce the shunt's R-to-L component if necessary.
- To restore the interventricular septum’s position (convex in the RV) and assistance to right-side ejection provided by LV contraction. An increase in left-side afterload is required to raise the pressure in the LV and cause the septum to bulge into the RV during systole.
- To maintain coronary perfusion of the RV as it can only rely on the systolic component of the coronary blood flow if PAP is close to MAP [45].
- Dobutamine (Dobutrex®): by increasing cAMP (ß1 stimulation), it induces significant pulmonary vasodilation in addition to its cardiostimulatory effect.
- Inodilators: phosphodiesterase-3 inhibitors (milrinone) increase cAMP (positive inotropic effect by slowing its degradation) while also lowering PAP and PVR (Corotrop® 0.5-0.75 mcg/kg/min).
- Epinephrin + milrinone is generally the most effective combination as it balances inotropic, pulmonary vasodilator, and systemic vasoconstrictor effects.
- Levosimendan (initial dose 6 mcg/kg, infusion 0.05-0.2 mcg/kg/min): this reduces PAP (right-side afterload) and increases ventricular contractility by sensitising troponin to Ca2+. It is the only inotropic agent that reduces mortality [37].

Possible surgical procedures include reopening or non-closure of the pericardium and sternum to decompress the RV in order to reduce ventricular interdependence. A Goretex™ patch is stitched to the edges of the cutaneous incision to insulate the heart pending secondary closure, which occurs
2-3 days later. ECMO and/or a right ventricular assist device are required in cases of pulmonary hypertensive crisis coupled with right-side failure.
Nitroglycerin has a slight pulmonary vasodilator effect, but mainly reduces RV preload and end-diastolic volume. It is only indicated for congenital heart patients with congestive failure of the RV (e.g. massive regurgitation of the pulmonary valve) as the reduced preload lowers the volume ejected by the RV if it is hypertrophied. Since the pulmonary vessels have virtually no α1 receptors, arteriolar vasodilators (sodium nitroprusside, phentolamine) mainly have a systemic effect and are not useful for selective treatment of PAH. Their effect is catastrophic in subjects with an R-to-L shunt or whose pulmonary blood flow is supplied from the systemic blood flow (Blalock-Taussig shunt, aortopulmonary collaterals). The recommended treatment for persistent PAH in neonates is inhaled NO combined with sildenafil and/or prostaglandins. Milrinone is indicated in cases of ventricular failure and ECMO in treatment-resistant cases [1].
Nitric oxide (NO•)
NO• (nitric oxide) is a physiological transmitter secreted by the endothelial cells that causes vascular muscle relaxation. It is equivalent to the substance known as EDRF (endothelium-derived relaxing factor). The half-life of NO• in the body as a gas is very short: 5-10 seconds [35]. NO• is permanently produced in the pulmonary circulation by NOS (nitric oxide synthase) from arginine. It facilitates cGMP synthesis, which slows the release of intracellular Ca2+. This maintains active vasodilation in perialveolar resistance vessels with small diameters. Its production is activated by arterial pulsatility. Hypoxia inhibits its activity and causes local vasoconstriction [13]. An intact endothelium is essential in order for such vasodilation to work – substances such as acetylcholine, bradykinin and histamine have a dilating effect on normal vessels but are vasoconstrictive if endothelial lesions are present. Similarly, substances that are normally vasodilatory have a powerful vasoconstrictive effect in subjects with severe respiratory diseases, lung transplants, chronic pulmonary hypertension or after extended CPB [14]. NO• is administered into the ventilator's inspiratory circuit (INOvent™ system) or an air-tight face mask if the child is on spontaneous ventilation. Its very high affinity for haemoglobin, with which it forms methaemoglobin, prompts its immediate inactivation once solubilised in the blood. Therefore it has no systemic effect when administered via the airways [35]. Methaemoglobin levels do not exceed 4-5% at FiNO of 30-40 ppm [12]. NO• can be toxic due to its instability – in the presence of O2, it oxidises quickly into NO2 and peroxynitrites in proportion to the quantity of NO• present, FiO2, and the contact time between the two. Humans can safely inhale a mean of 25 ppm of NO•. For periods of several tens of hours, this value may rise to 80 ppm [38].
With 5 to 20 ppm of NO• in inhaled gases, it is possible to reverse non-fixed pulmonary vasoconstriction without any systemic vasodilation. PVR falls by 10 to 37% according to literature [33]. Its directly dose-dependent effect takes 1 to 3 minutes for onset, but quickly dissipates after the NO• is discontinued. However, it is not possible to reduce PVR below its baseline value in normal children. Results show that NO• is an effective means of addressing hypoxic pulmonary vasoconstriction in healthy subjects [19] or thromboxane release through the reaction of heparin and protamine [18]. However, pulmonary reactivity is low if the pulmonary vessels are anomalous and fibrotic. In contrast to animal tests in which the values used go up to 1,000 ppm, concentrations of over 40 ppm probably offer little benefit in humans [38]. Although NO• counteracts hypoxic vasoconstriction of ventilated areas, it cannot access unventilated areas, which remain hypoperfused. When administered to subjects with respiratory distress syndrome, it therefore improves gas exchange by reducing the shunt effect, contrary to conventional vasodilators [16].
Some pathologies appear to respond to low concentrations (5-10 ppm), while other situations require high concentrations (20-40 ppm). In children, inhalation of NO• only reduces the pulmonary vascular resistance of the small perialveolar vessels. It does not lead to any systemic modifications or dilation of the ductus arteriosus. The usually admitted indications are the following [12].
- Neonatal pulmonary hypertension [40];
- Pulmonary hypertensive crises in subjects with congenital heart diseases [24,27];
- RV rescue in the event of an acute increase in afterload;
- RV failure following heart transplant or left ventricular assist device placement [30];
- Preoperative test of PAH reversibility [46];
- Withdrawal from ECMO.
Despite its benefits, use of NO• is limited by several factors [12,34].
- Platelet adhesiveness is inhibited;
- Hypertension reappears 3-5 minutes after NO• is discontinued; sudden discontinuation may cause a rebound effect and a PAH crisis;
- Low FiO2 is required;
- NO2 is produced;
- The equipment can be a cumbersome addition to the inspiratory circuit, it may induce metal and plastic corrosion;
- NO2 and methaemoglobin formation must be monitored.
| Treatment of pulmonary hypertension |
|
Correction of acidosis, hypothermia, anaemia, stress and pain. Deepening of anaesthesia
(fentanil, curare). Ventilation and inhalation (no systemic effect) - Hyperventilation with low intrathoracic pressure - NO• (10-30 ppm) - Nebulised prostacyclin (iloprost, treprostinil), milrinone or nitroglycerin Pulmonary vasodilators (risk of systemic hypotension) - Prostacyclins (epoprostenol, treprostinil) - Phosphodiesterase-3 inhibitors (milrinone) - Phosphodiesterase-5 inhibitors (sildenafil) - Cardiac stimulants: milrinone + epinephrin, dobutamine, levosimendan - Less effective: nitroglycerin, nesiritide, riociguat, Mg2+ - Calcium channel blockers contraindicated if systemic vasodilation presents a risk of R-to-L shunting Systemic vasoconstrictors - Norepinephrin - Vasopressin Chronic treatment - Endothelin receptor antagonists (bosentan, ambrisantan) - Phosphodiesterase-5 inhibitors (sildenafil, tadalafil) - Prostacyclins (epoprostenol, treprostinil, iloprost) Non-drug procedures - Non-closure or reopening of the pericardium - ECMO, right ventricular assist device - Atrial septostomy - Potts shunt - Lung or heart-lung transplant The key to improving haemodynamics lies not only in lowering PAP, but also in lowering CVP and increasing systolic volume as a result of improved RV function. |
Anaesthesia and PAH
Anaesthetic agents have little impact on pulmonary circulation, except ketamine, N2O and desflurane, which cause sympathetic stimulation and pulmonary hypertension [15]. Recommendations for anaesthetising children with PAH may be set out as follows [17].
Anaesthetic agents have little impact on pulmonary circulation, except ketamine, N2O and desflurane, which cause sympathetic stimulation and pulmonary hypertension [15]. Recommendations for anaesthetising children with PAH may be set out as follows [17].
- Deep anaesthesia (stress-free) using fentanil and a hypnotic agent (halogenated agent, propofol) to limit the risk of PAH crises.
- Hyperventilation with low intrathoracic pressure (IPPV): respiratory alkalosis (FiO2 1.0, PaCO2 30-35 mmHg, pH 7.5); minimum PEEP to prevent atelectasis; avoid hypoventilation and acidosis.
- Normothermia.
- Maintain preload: avoid any hypovolaemia.
- Maintain SVR (vasoconstrictor): ensure satisfactory coronary perfusion pressure, reduce the shunt's R-to-L component, reposition the interventricular septum to the right (TEE examination).
- In the event of moderate PAH with a strictly L-to-R shunt, attempts should be made to reduce the SVR to avoid exacerbating the shunt.
- In cases of PAH with a bidirectional shunt, the Qp/Qs ratio should be balanced to the greatest possible extent (ideally Qp = Qs) by preventing any changes to vascular resistance.
- Monitoring: an arterial line and CVP monitoring are necessary. A system for measuring cardiac output (Swan-Ganz, PiCCO) is also beneficial in major operations. However it is difficult to fit such catheters due to young children's size. Moreover, a pulmonary catheter may be impossible to position or even dangerous in some anatomical configurations.
- Transfusion threshold: Hb 100 g/L.
Among children with PAH, the incidence of severe complications (PAH crisis, death) in noncardiac surgery is 2-3%. This risk increases 8 times (OR = 8.1) among those with Eisenmenger's syndrome (systemic or suprasystemic PAP) [9].
Ventilation and pulmonary hypertension
Ventilation involves a compromise between active hyperventilation and maintaining low mean intrathoracic pressure (ITP). If the tidal volume is low, there is a risk of atelectasis, hypercapnia and increased PVR in the small perialveolar vessels. If it is high, ITP rises and the large extra-alveolar vessels are compressed due to hyperinflation [17]. Ideally, the tidal volume should be the same as the functional residual capacity (FRC) (Figure 14.19).
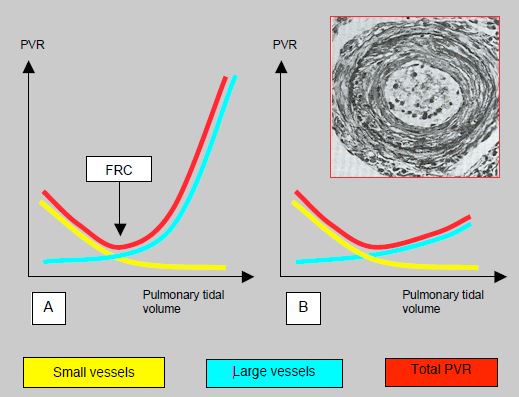
Figure 14.19: Trend for pulmonary vascular resistance (PVR) with ventilation. A: Normal situation. At low tidal volume, PVR is raised in the small vessels by hypoventilation and hypercapnia. At high tidal volume, it is raised in the large perialveolar vessels, which are mechanically stretched and compressed. The resultant (red curve) shows that the best compromise is reached at the volume of the functional residual capacity (FRC). This represents ventilation at 6-8 mL/kg [17]. B: In subjects with chronic pulmonary hypertension, the artery walls are thickened with increased tunica media musculature, subendothelial proliferation, and fibrosis of the tunica externa (see pulmonary artery cross-section in the inset). The intrathoracic pressure is no longer capable of compressing these vessels. The curve representing the large vessels is therefore flattened and total PVR is no longer U-shaped. It no longer rises at high tidal volume. Therefore, these patients tolerate hyperventilation under IPPV very well.
Adjustments must be made to the tidal volume, rate and ventilation method (pressure-controlled ventilation) to achieve the lowest possible PaCO2 and mean ITP. To that end, shortening the duration of inspirium reduces the mean ITP more than reducing the peak inspiratory pressure value. An I:E ratio 1:2 should be sought.
Adjustments must be made to the tidal volume, rate and ventilation method (pressure-controlled ventilation) to achive the lowest possible PaCO2 and mean ITP. To that purpose, shortening the duration of inspirium reduces the mean ITP more than reducing the peak inspiratory pressure value. An I:E ration < 1:2 should be sought for.
Concerns over the negative impact of general anaesthesia on ventilation are unfounded for three reasons.
Ventilation and pulmonary hypertension
Ventilation involves a compromise between active hyperventilation and maintaining low mean intrathoracic pressure (ITP). If the tidal volume is low, there is a risk of atelectasis, hypercapnia and increased PVR in the small perialveolar vessels. If it is high, ITP rises and the large extra-alveolar vessels are compressed due to hyperinflation [17]. Ideally, the tidal volume should be the same as the functional residual capacity (FRC) (Figure 14.19).
Figure 14.19: Trend for pulmonary vascular resistance (PVR) with ventilation. A: Normal situation. At low tidal volume, PVR is raised in the small vessels by hypoventilation and hypercapnia. At high tidal volume, it is raised in the large perialveolar vessels, which are mechanically stretched and compressed. The resultant (red curve) shows that the best compromise is reached at the volume of the functional residual capacity (FRC). This represents ventilation at 6-8 mL/kg [17]. B: In subjects with chronic pulmonary hypertension, the artery walls are thickened with increased tunica media musculature, subendothelial proliferation, and fibrosis of the tunica externa (see pulmonary artery cross-section in the inset). The intrathoracic pressure is no longer capable of compressing these vessels. The curve representing the large vessels is therefore flattened and total PVR is no longer U-shaped. It no longer rises at high tidal volume. Therefore, these patients tolerate hyperventilation under IPPV very well.
Adjustments must be made to the tidal volume, rate and ventilation method (pressure-controlled ventilation) to achieve the lowest possible PaCO2 and mean ITP. To that end, shortening the duration of inspirium reduces the mean ITP more than reducing the peak inspiratory pressure value. An I:E ratio 1:2 should be sought.
Adjustments must be made to the tidal volume, rate and ventilation method (pressure-controlled ventilation) to achive the lowest possible PaCO2 and mean ITP. To that purpose, shortening the duration of inspirium reduces the mean ITP more than reducing the peak inspiratory pressure value. An I:E ration < 1:2 should be sought for.
Concerns over the negative impact of general anaesthesia on ventilation are unfounded for three reasons.
- The increase in afterload for the RV induced by IPPV is very small compared to its normal afterload – adding mean ITP of 10 mmHg to PAPmean of 50 mmHg changes haemodynamic conditions less than if PAPmean is normal (20 mmHg).
- The left side of the graph in Figure 14.19 does not actually apply to children with PAH since the thickened, rigid walls of their pulmonary vessels prevent any compression by high tidal volume ventilation.
- With IPPV, it is possible to hyperventilate patients and thus reduce their PVR. In contrast, arterial vasodilation prompted by neuraxial anaesthesia would exacerbate the shunt's R-to-L component, causing arterial desaturation, and increasing cyanosis [3].
There is therefore no reason to fear an additional increase in PAP if mechanical hyperventilation is used.
| Anaesthesia and PAH |
|
Avoid any increase in PVR due to: hypoventilation, hypercapnia, atelectasis, high intrathoracic pressure, acidosis, hypothermia, sympathetic stimulation, stress, pain
Anaesthesia: - Deepening anaesthesia and analgesia - Hyperventilation with low intrathoracic pressure, respiratory alkalosis, normoxia, NO - Maintain preload - Maintain SVR (systemic vasoconstrictor) - Normothermia. In cases of chronic PAH, IPPV with low intrathoracic pressure is well tolerated for 3 reasons: - The increase in right-side afterload is low compared to the chronically high PAP - The hypertrophied RV can withstand increased afterload - Thickened and fibrotic pulmonary vessels are not compressible by IPPV |
© BETTEX D, BOEGLI Y, CHASSOT PG, June 2008, last update February 2020
References
- ABMAN SH, HANSMANN G, ARCHER SL, et al. Pediatric pulmonary hypertension. Guidelines from the American Heart Association and American Thoracic Society. Circulation 2015; 132:2037-99
- ALTHOFF TF, KNEBEL P, PANDA A, et al. Long-term follow-up of a fenestrated Amplatzer atrial septal occluder in pulmonary arterial hypertension. Chest 2008; 133:283-5
- AMMASH NA, CONNOLLY HM, ABEL MD, WARNES CA. Noncardiac surgery in Eisenmenger syndrome. J Am Coll Cardiol 1999; 33:222-7
- BARR FE, MACRAE D, Inhaled nitric oxide and related therapies. Pediatr Crit Care Med 2010; 11(Suppl): S30-S36
- BARUTEAU AE, et al. Potts shunt in children with idiopathic pulmonary arterial hypertension: long-term results. Ann Thorac Surg 2012; 94:817-24
- BEGHETTI M, BERGER RM, SCHULZE-NEICK I, et al. Diagnostic evaluation of paediatric pulmonary hypertension in current clinical practice. Eur Respir J 2013; 42:689-700
- BLAISE G, LANGLEBEN D, HUBERT B. Pulmonary arterial hypertension. Pathophysiology and anesthetic approach. Anesthesiology 2003; 99:1415-32
- BUNDTS W, VAN PELT N, GILLYNS H, et al. Residual pulmonary vasoreactivity to inhaled nitric oxide in patients with severe obstructive pulmonary hypertension and Eisenmenger syndrome. Heart 2001; 86:553-9
- CARMOSINO MJ, FRIESEN RH, DORAN A, IVY DD : Perioperative complications in chidren with pulmonary hypertension undergoing noncardiac surgery or cardiac catheterization. Anesth Analg 2007 ; 104 :521-7
- CERRO MJ, ABMAN S, DIAZ G, et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular diasease : report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ 2011 ; 1 :286-98
- CHASSOT PG, BETTEX DA. Anesthesia and adult congenital heart disease. J Cardiothorac Vasc Anesth 2006; 20:414-37
- CHECCHIA PA, BRONICKI RA, GOLDSTEIN B. Review of inhaled nitric oxide in the pediatric cardiac surgery stting. Pediatr Cardiol 2012; 33:493-505
- CREMONA D, DINH XUANG AT, HIGGENBOTHAM T. Endothelium-derived relaxing factor and the pulmonary circulation. Lung, 1991; 169:185-202
- DINH XUAN AT, HIGGENBOTTAM TW, CLELLAND CA, et al. Impairment of endothelium-dependent pulmonary artery relaxation in chronic obstructive lung disease. N Engl J Med 1991; 324:1539-47
- EBERT TJ, MUZI M. Sympathetic hyperactivity during desflurane anesthesia in healthy volunteers: a comparison with isoflurane. Anesthesiology 1993; 79:444-53
- FALKE K, ROSSAINT R, PISON U, et al. Inhaled nitric oxide selctively reduces pulmonary hypertension in severe ARDS and improves gaz exchange as well as right heart ejection fraction. Am Rev Respir Dis 1991; 143 (suppl):A248
- FISCHER LG, VAN HAKEN H, BÜRKLE H. Management of pulmonary hypertension: Physiological and pharmacological considerations for anesthesiologists. Anesth Analg 2003; 96:1603-16
- FRATTACCI M, FROSTELL C, CHEN TY, et al. Inhaled nitric oxide. A selective vasodilator of heparin-protamine vasoconstriction in sheep. Anesthesiology 1991, 75:990-9
- FROSTELL C, BLOMQVIST H, HEDENSTIERNA G, et al. Inhaled nitric oxide selectively reverse human hypoxic pulmonary vasoconstriction without causing systemic vasodilatation. Anesthesiology 1993; 78:427-35
- GALIÉ N, HUMBERT M, VACHIERY JL, et al. 2015 ESC/ERS Guideline for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2016; 37:67-119
- GALLE N, HOEPER MM, HUMBERT M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2009; 30:2493-537
- GHOFRANI HA, WIEDEMAN R, ROSE F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension. A randomized controlled trial, Lancet 2002; 360:895-900
- GUAZZI M, BORLAUG BA. Pulmonary hypertension due to left heart disease. Circulation 2012; 126:975-90
- HAYDAR A, MAURIAT P, POUARD P, et al. Inhaled nitric oxide for postoperative pulmonary hypertension in patients with congenital heart defects. Lancet 1992; 340:1545
- IVY DD. Advances in pediatric pulmonary arterial hypertension. Curr Opin Cardiol 2012; 27:70-81
- IVY DD, ABMAN SH, BARST RJ, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol 2013; 62:D117-26
- KADOSAKI M, KAWAMURA T, OYAMA K, et al. Usefulness of nitric oxide treatment for pulmonary hypertensive infants during cardiac anesthesia. Anesthesiology 2002; 96:835-40
- KASSEM E, HUMPL T, FRIEDBERG MK. Prognostic significance of 2-dimensional, M-mode, and Doppler echo indices of right ventricular function in children with pulmonary arterial hypertension. Am Heart J 2013; 165:1024-31
- KERBAUL F, RONDELET B, COLLART F, et al. Hypertension artérielle pulmonaire en anesthésie-réanimation. Ann Fr Anesth Réanim 2005; 24:528-40
- KIELER-JENSEN N, LUNDIN S, RICKSTEN S. Vasodilator therapy after heart transplantation: effects of inhaled nitric oxide and intravenous prostacyclin, prostaglandin E1, sodium nitroprusside. J Heart Lung Transplant 1995; 14:436-43
- LATUS H, DELHAAS T, SCHRANZ D, APITZ C. Treatment of pulmonary arterial hypertension in children. Nat Rev Cardiol 2015; 12:244-54
- MICHELAKIS E, TYMCHAK W, LIEN D, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: Comparison with inhaled nitric oxide. Circulation 2002; 105:2398-403
- MILLER O, CELERMEYER D, DEANFIELD J, et al. Very low dose inhaled nitric oxide: A selective pulmonary vasodilator after operations for congenital heart disease. J Thorac Cardiovasc Surg 1994; 108:487-94
- MILLER O, TANG S, KEECH A, et al. Rebound pulmonary hypertension on withdrawal from inhaled nitric oxide. Lancet 1995; 346:51-2
- MONCADA S, PALMER R, HIGGS E. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacological Review 1991; 43:109-42
- OECHSLIN E. Eisenmenger's syndrome. In: GATZOULIS MA, WEBB GD, DAUBENEY PEF, Eds. Diagnosis and management of adult congenital heart disease. Edinburgh, Churchill Livingstone 2003, pp 363-77
- POLLESELLO P, PARISSIS J, KIVIKKO M, et al. Levosimendan meta-analyses: is there a pattern in the effect on mortality ? Int J Cardiol 2016; 209: 77-83
- RICH GF, JOHNS RA. Nitric oxide and the pulmonary circulation. Advances in Anaesthesia 1994; 11:1-25
- RICH S, MCLAUGHLIN VV. The effects of chronic prostacyclin therapy on cardiac output and symptoms in primary pulomonary hypertension. J Am Coll Cardiol 1999; 34:1184-7
- ROBERTS JD, POLANDER D, LANG P, et al. Inhaled nitric oxide in persistant pulmonary hypertension in the newborn. Lancet 1992; 340:818-9
- RUBIN LJ, BADESCH DB, BARST RJ, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002; 346:896-903
- SCHROEDER RA, WOOD GL, PLOTKIN JS, et al. Intraoperative use of inhaled PGI(2) for acute pulmonary hypertension and right ventricular failure. Anesth Analg 2000; 91:291-5
- SIMONEAU G, GATZOULIS MA, ADAIA I,, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D34-41
- TAKATSUKI S, NAKAYAMA T, JONE PN, et al. Tissue Doppler imaging predicts adverse outcome in children with idiopathic pulmonary arterial hypertension. J Pediatr 2012; 161:1126-31
- THUNBERG CA, GAITAN BD, GREWAL A, et al. Pulmonary hypertension in patients undergoing cardiac surgery: pathophysiology, perioperative management and outcome. J Cardiothorac Vasc Anesth 2013; 27: 551-72
- TURANLAHTI M, LAITINEN P, PESONEN E. Preoperative and postoperative response to inhaled nitric oxide. Scand Cardiovasc J 2000; 34:46-52
- VALAPOUR M, et al. OPTIN/SRTR 2011 annual data report: lung. Am J Transplant 2013; 13 (suppl 1):149-77
- VAN LOON RL, ROOFTHOOFT MT, HILLEGE HL, et al. Pediatric pulmonary hypertension in the Netherlands: epidemiology and characterization during the perios 1991 to 2005. Circulation 2011; 124:1755-64