Congenital heart disease can raise or lower the pulmonary blood flow (Qp) (Table 14.6). The increase in pulmonary blood flow by a L-to-R ventricular or arterial shunt (VSD, atrioventricular canal defect, truncus arteriosus, surgical aortopulmonary shunt, transposition of the great arteries + VSD) causes a gradual rise of pulmonary arterial resistance (PVR), secondarily responsible for hypertrophy of the RV. Pulmonary arterial hypertension (PAH) is defined by a mean PAP > 25 mmHg at rest, or pulmonary vascular resistance (PVR) > 300 dynes•cm•s-5 (> 3 U Wood/m2) [3,5]. Since the PAP of neonates takes approximately 2 months to drop to its normal adult value, pulmonary hypertension is determined as such only from 3 months of age [7]. It occurs in 50% of cases of VSD and patent ductus arteriosus, where both volume and pressure overload are present, but in only 10% of cases of ASD, where volume overload only is concerned. It appears in infancy in cases of VSD, but only in adulthood in cases of ASD (see Paediatric Pulmonary Hypertension) [8].
Vascular wall shear stress caused by excessive pulmonary blood flow prevents normalisation of PVR during the first months of life. Continued high pulmonary blood flow and excessive PAP lead to a hypertrophy of the media in most proximal arteries and an expansion of the smooth muscle in peripheral vessels that are normally not muscular. It gradually leads to intimal proliferation, plexiform lesions and adventitial fibrosis, as well as an inflammatory reaction and disseminated thrombosis (Figure 14.12) [9,10].
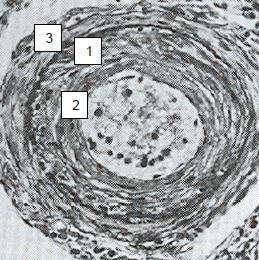
Figure 14.12 : Image anatomo-pathologique d’une artériole pulmonaire en cas de syndrome d’Eisenmenger. 1 : hypertrophie de la média. 2 : prolifération intimale. 3 : fibrose [10]. La paroi d’une telle artère ne peut plus être collabée par les pressions ventilatoires en ventilation mécanique.
Finally, arborisation is suppressed and the number of distal vessels is reduced: the PAH is fixed and irreversible. This leads to Eisenmenger’s syndrome, characterised by a mean PAP > 50 mmHg and pulmonary vascular resistance > 800 dynes•cm•s-5. The PAP is such that the flow through the L-to-R shunt is then bidirectional or reversed (suprasystemic PAP). Faced with this pressure overload, the RV undergoes massive hypertrophy and thickens considerably. The mass of the two ventricles is equal during intrauterine life; the RV retains its foetal structure if it is exposed to a high afterload at birth and its hypertrophy occurs in parallel to the development of the LV [4]. The RV is more resistant to an increase in afterload during childhood than when this occurs secondarily during adulthood. Outcomes for children with congenital heart disease are better than for children with primary idiopathic PAH and for adults with drug-induced or thromboembolic PAH [2],
- ASD: 90% of 40-year survival;
- Eisenmenger's syndrome. 80% of 10-year survival;
- Primary idiopathic PAH: 66% of 5-year survival;
- Thromboembolic disease: 35% of 5-year survival;
The prognosis for PAH is closely related to right heart function. It is poor if the RV fails and dilates. Indeed, PAH is linked to the RV’s ability to chronically generate high pulmonary pressure. Once the RV fails, PAP tends to fall again, while the haemodynamic situation worsens and SVR continues to rise. Increased pulmonary blood flow and compression of the airways by dilated and swollen vessels lead to a decrease in lung compliance, increased airway resistance and increased respiratory effort. The severity of these consequences determines operability and risks associated with the postoperative period. If surgical correction is performed early, the PAH gradually fades. However, if the PVR/SVR ratio is already > 0.4 (mPAP/MAP > 0.7), surgery is contraindicated [1]. Down syndrome is closely associated with PAH, not only in connection with patent ductus arteriosus, but also in relation to sleep apnoea syndrome, pulmonary aspiration, bronchoaspiration and gastroesophageal reflux disease, which are common amongst patients with trisomy 21 [11].
| Pulmonary arterial hypertension (PAH) |
| Definition of PAH: mean PAP > 25 mmHg at rest Eisenmenger syndrome (irreversible PAH): mean PAP > 50 mmHg, PVR > 800 dynes•cm•s-5 and bidirectional shunt with cyanosis (L-to-R shunt + R-to-L component) Operability criteria: PVR/SVR ratio < 0.4 The prognosis of PAH is closely related to right heart function. It is poor if the RV fails and dilates |
Echocardiographic calculation of PAP
A pulmonary catheter is not required for measuring PA pressure. A reliable estimate is possible using echocardiography, provided that the patient presents with tricuspid insufficiency (TI). This occurs if right-sided pressure increases, i.e. in almost all patients with pulmonary hypertension. The maximum velocity of the systolic TI jet is dependent on the pressure gradient prevailing during systole between the RV and RA. The simplified Bernoulli equation establishes the relationship between velocity and pressure:
∆P = 4 (Vmax)2
where ∆P is the pressure gradient between RV and RA during systole and Vmax the maximum TI velocity.
RA pressure is estimated (mean value of 10 mmHg) or measured by CVP. If there are no lesions in the right ventricular outflow tract or in the pulmonary valve, the systolic pressure of the RV equals the systolic pressure of the PA. RA pressure must be added to the value of the RV - RA gradient to calculate the value of PsystRV. This changes the equation as follows:
sPAP = 4 (Vmax TI)2 + RAP
In echocardiography, the Doppler beam is aligned with the TI jet and the computer automatically performs the calculation (see Figure 15.7). However, this calculation only correlates to a limited extent with direct measurement of systolic pulmonary arterial pressure (sPAP) by a pulmonary artery catheter (r = 0.7) [12]. Among patients with congenital heart diseases, this calculation may be distorted by two situations [6]:
- Stenosis of the RV outflow tract; the RV systolic pressure is not equal to the sPAP;
- Presence of a VSD; RV pressure is contaminated by LV pressure.
If a VSD is present, it is possible to calculate RV systolic pressure on the basis of the velocity of the blood flow in the L-to-R shunt. The maximum shunt velocity (Vmax VSD) is used to measure the difference of pressure during systole between the LV and RV. The subtraction of Vmax VSD from systemic arterial pressure in systole APsyst (assuming no aortic pathology) gives RV systolic pressure:
PsystRV = APsyst - 4 (Vmax VSD)2
| Calculation of systolic pulmonary pressure |
| At echocardiography: PAPsyst = 4 (Vmax TI)2 + RAP (only if the RV outflow tract is normal) In the case of VSD: Psyst RV = APsyst - 4 (Vmax VSD)2 |
© BETTEX D, BOEGLI Y, CHASSOT PG, June 2008, last update February 2020
References
- ABMAN SH, HANSMANN G, ARCHER SL, et al. Pediatric pulmonary hypertension. Guidelines from the American Heart Association and American Thoracic Society. Circulation 2015; 132:2037-99
- BENZA RL, MILLER DP, BARST RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from REVEAL. Chest 2012; 142:448-56
- BLAISE G, LANGLEBEN D, HUBERT B. Pulmonary arterial hypertension. Pathophysiology and anesthetic approach. Anesthesiology 2003; 99:1415-32
- BRONICKI RA, BADEN HP. Pathophysiology of right ventricular failure in pulmonary hypertension. Pediatr Crit Care Med 2010; 11(Suppl):S15-S22
- CERRO MJ, ABMAN S, DIAZ G, et al. A consensus approach to the classification of pediatric pulmonary hypertensive vascular diasease : report from the PVRI Pediatric Taskforce, Panama 2011. Pulm Circ 2011 ; 1 :286-98
- CHASSOT PG, BETTEX DA. Anesthesia and adult congenital heart disease. J Cardiothorac Vasc Anesth 2006; 20:414-37
- IVY DD, ABMAN SH, BARST RJ, et al. Pediatric pulmonary hypertension. J Am Coll Cardiol 2013; 62:D117-26
- OECHSLIN E. Eisenmenger's syndrome. In: GATZOULIS MA, WEBB GD, DAUBENEY PEF, Eds. Diagnosis and management of adult congenital heart disease. Edinburgh, Churchill Livingstone 2003, pp 363-77
- PALEVSKY HI, SCHLOO BL, PIETRA CC, et al. Primary pulmonary hypertension: Vascular structure, morphometry, and responsiveness to vasodilator agents. Circulation 1989; 80:1207-21
- RABINOVITCH M, HAWORTH SG, CASTANEDA AR, et al. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation 1978; 58:1007-22
- RIGBY ML, ROSENTHAL M. Cardiorespiratory interactions in paediatrics. "It's (almost always) the circulation stupid!" Paed Respir Rev 2017; 22:60-5
- THUNBERG CA, GAITAN BD, GREWAL A, et al. Pulmonary hypertension in patients undergoing cardiac surgery: pathophysiology, perioperative management and outcome. J Cardiothorac Vasc Anesth 2013; 27: 551-72