When valvular disease leads to a reduction in effective systolic flow, the heart has three compensatory mechanisms.
- Increased sympathoadrenergic activity (alpha and beta catecholamines, angiotensin II, aldosterone, ADH): Arterial vasoconstriction and tachycardia help to maintain cardiac output. The patient's resting blood pressure and heart rate are probably the best possible haemodynamic compromise in their situation.
- Frank-Starling effect: increase in preload and circulating volume (water and salt retention). Dilatation makes the ventricle more spherical, which is pathological but has a temporary mechanical advantage because the circumferential shortening of the fibres to eject the same volume is reduced. This mechanism is mainly used in acute lesions or regurgitation. The upstream effects are an increase in atrial pressure and congestion (peripheral or pulmonary).
- Hypertrophy: upstream of a valvular stenosis, the ventricle enlarges to ensure blood propulsion despite the obstacle to ejection (concentric hypertrophy of the LV in aortic stenosis); downstream, the ventricle is reduced in size (small LV in mitral stenosis). Valvular insufficiency also affects the size of the two upstream and downstream chambers: the ventricle must receive an increased volume of blood with each beat (dilation) and increase its systolic volume to maintain anterograde flow (eccentric hypertrophy); the chamber receiving the regurgitation also dilates (dilated atrium in MI or TI).
Functional anatomy, as revealed by TEE in a 4-chamber view, is an excellent illustration of the remodelling and compensations that the body makes to maintain haemodynamic balance. It shows the dominant stresses and their consequences. Pressure overload (aortic stenosis) causes concentric left ventricular hypertrophy, resulting in a thickened LV with a small cavity, whereas volume overload (mitral or aortic regurgitation) causes dilatation of the ventricular cavity and left atrium. Mitral stenosis results in a small left ventricle and a large left atrium (see Figure 11.14). In pulmonary hypertension, the right ventricle becomes enlarged and dilated, and the progressive development of tricuspid regurgitation enlarges the right atrium, with the septum tilting to the left (see Figure 11.16).
Ventricular hypertrophy
The number of heart muscle cells doubles during the first month of life and then remains stable. The lifespan of these cells is therefore the same as that of the individual, except for those that degenerate and are replaced by fibrocytes. Since they cannot proliferate, cardiomyocytes replicate the sarcomeric contractile units in series (eccentric hypertrophy) or in parallel (concentric hypertrophy). Growth hormone, insulin and angiotensin II are the main anabolic factors that stimulate mRNA transcription and protein synthesis; in addition to cell growth, these substances are responsible for hypertrophy and hyperplasia of cardiac cells (myocytes and fibroblasts) in response to pressure or volume overload [2]. Recent research has also identified a whole cascade of elements involved in the genesis of hypertrophy: NO, thyroid hormone, catecholamines, insulin, neuroregulin, integrin, natriuretic peptides, as well as several intracellular signalling systems, epigenetic pathways and fetal genes or genes encoding calcium metabolism [7]. In hypertrophy, non-muscular cells (vascular, interstitial, etc.) may undergo concomitant hyperplasia [9], but the density of capillaries per unit mass is reduced [10]. Converting enzyme inhibitors and angiotensin II or renin antagonists slow down this ventricular remodelling.
A distinction is usually made between physiological and pathological hypertrophy. The former is induced by exercise or pregnancy; it increases cardiac mass by 10-20%, improves contractile function and is reversible. The latter is caused by arterial hypertension, aortic stenosis, volume overload (valvular insufficiency), myocardial ischaemia and certain metabolic or genetic disorders (cardiomyopathy). Often associated with obesity and diabetes, it leads to impaired contractile function, secondary ventricular dilatation and systolic-diastolic heart failure. It is associated with interstitial and perivascular fibrosis, fibroblastic proliferation and increased collagen deposition [7].
Pressure overload (valvular stenosis) leads to increased systolic wall stress and concentric hypertrophy with narrowing of the ventricular cavity; the mechanism is parallel replication of the sarcomeres. Chronic volume overload (valvular insufficiency) leads to an increase in diastolic wall stress, resulting in eccentric hypertrophy with dilatation of the ventricular cavity and serial replication of the sarcomeres.
Whether pressure overload (aortic stenosis) or volume overload (aortic or mitral regurgitation), left ventricular hypertrophy develops in three stages, which can be analysed according to Laplace's law, which defines wall stress (σ ): σ = (P - r) / 2h (Figure 11.18) [6,9].
- The work demanded exceeds the normal capacity of the myocardium: wall stress increases by increasing pressure P (resistance to ejection) or radius r (dilatation).
- Hypertrophy (increase in thickness h) normalises the wall tension as the wall thickens; the size of the ventricular cavity decreases (pressure overload) or increases (volume overload). The critical limit for myocardial mass is 500 g or 300 g/m2 (normal heart weight: 250 g).
- Decompensation: The physiological mechanisms are overwhelmed and wall stress increases again due to pressure overload (excess P) or volume overload (increase r). The predominant symptom is congestive heart failure.
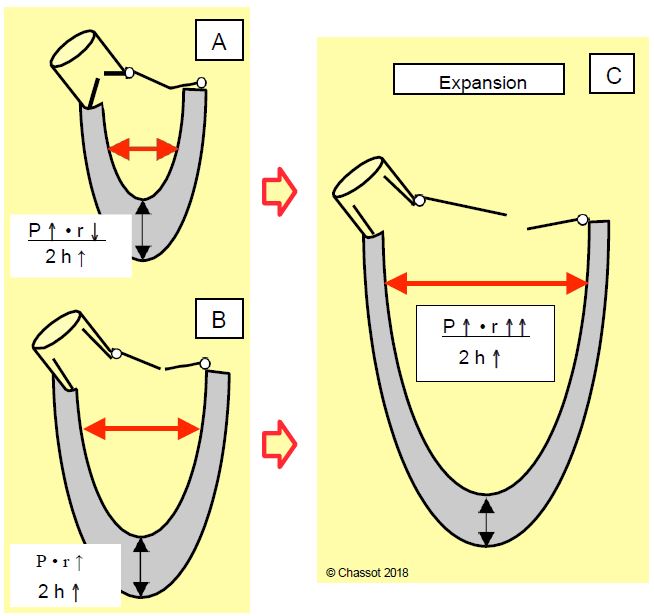
Figure 11.18: Left ventricular hypertrophy (LVH) and Laplace's law: wall stress is equal to the product of pressure and radius divided by 2 times thickness. A: Concentric LVH (pressure overload). The increase in pressure (P) is compensated by a decrease in the radius (r) of the cavity and an increase in the thickness (h) of the ventricular wall. The wall stress is stable. B: Eccentric or dilatative LVH (volume overload). The radius increases without an increase in pressure, which is compensated by an increase in wall thickness. C: Decompensation phase. LV pressure and size increase beyond what can be compensated by physiological mechanisms.
During pressure overload, wall thickness and internal work (isometric contraction) are greater than during volume overload: the ratio of ejection work to total work (pressure + ejection) is lower, so the efficiency of the ventricular motor has decreased: mVO2 increases to provide the same ejection work. During concentric hypertrophy, the overall contractile performance of the myocardium is increased, but calculated as work done in relation to myocardial mass, it is below the norm [5]. On the other hand, the diastolic function of this thickened wall is always impaired. This negative lusitropic effect is very pronounced in concentric hypertrophy, but is also present in dilatative hypertrophy, despite the fact that the ventricle is able to accommodate large volumes. Indices of diastolic relaxation and distensibility decrease as ventricular mass increases. The associated fibrosis, which is irreversible, contributes to this significant decrease in compliance [11]. Diastolic dysfunction manifests itself in two ways.
- A marked dependence of flow on preload volume, filling pressure and rate (duration of diastole).
- A progressive increase in filling pressures in the dilating atrium and upstream veins (pulmonary or hepatic stasis).
The concentric hypertrophy (LVH) characteristic of aortic stenosis is associated with myocardial ischaemia for several reasons [1,3,4,8].
- Degenerative, calcified aortic stenosis is part of the same underlying disease as hypertensive and hypercholesterolemic polyvasculopathy; as a result, the coronary tree is often affected and myocardial ischaemia is often associated.
- LVH increases basal metabolism and increases mVO2 by increasing myocardial mass; hypertrophied muscle consumes up to 50% more O2 to perform the same external work.
- Increased wall stress, which occurs when muscle thickness is out of proportion to diameter and afterload, is a major determinant of mVO2.
- At the same aortic diastolic pressure, the increase in intraventricular pressure due to the increase in afterload and diastolic dysfunction reduces the value of coronary perfusion pressure (CPP = PAdiastAo - PtdLV). In the event of arterial hypotension, CPP collapses because PAdiast in the aorta falls but Ptd in the LV does not change (aortic stenosis is fixed).
- Vascular hypertrophy and hyperplasia lag behind the increase in muscle mass: capillary density is reduced by 20-30% in concentric LVH.
- The subendocardial region is particularly vulnerable for two reasons: 1) the coronary vessels are crushed by the high Ptd of the LV, and 2) they are compressed by the excessive muscle mass on their longer way between the epicardium and the endocardium. Repeated ischaemic episodes lead to subendocardial fibrosis, which further reduces ventricular compliance.
- The Venturi effect caused in the sinuses of Valsalva by the high velocity of the systolic jet induces local depression and retrograde flow in the coronary trunks during systole.
| Adaptation to heart valve disease |
| Neurohumoral stimulation (catecholamines, angiotensin II, aldosterone, ADH).
Frank-Starling effect (increase in preload). Concentric ventricular hypertrophy: pressure overload (stenosis) - Preserved systolic function - Diastolic dysfunction - Greater dependence on preload - Dependence on sinus rhythm and frequency - Increased risk of myocardial ischaemia Eccentric ventricular hypertrophy: volume overload (insufficiency) - Ventricular dilatation - Systolic dysfunction LVH is a temporary compensatory measure that develops secondarily into dilatation and cardiac decompensation. |
© CHASSOT PG, BETTEX D, August 2011, last update November 2019
References
- BONOW RO, BRAUNWALD E. Valvular heart disease. In: ZIPES DP, et al, eds. Braunwald's heart disease. A textbook of cardiovascular medicine. 7th edition. Philadelphia: Elsevier Saunders, 2005, 1553-632
- CRACKOWER MA, et al. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature 2002; 417:822-8
- GHALI JK, LIAO Y, SIMMONS B, et al. The prognostic role of left ventricular hypertrophy in patients with and without coronary artery disease. Ann Intern Med 1992; 117:831-8
- HARRISON DG, BARNES DH, HIRAZKA LF, et al. The effect of cardiac hypertrophy on the coronary collateral circulation. Circulation 1985; 71:1135-41.
- KRAYENBÜHL HP, HESS OM, MONRAD ES, et al. Left ventricular myocardial structure in aortic valve disease before, intermediate and later after aortic valve replacement. Circulation 1989; 79:744-52
- MEERSON FZ. The failing heart. In: MEERSON FZ. Adaptation and deadaptation. New York, Raven Press, 1983
- NAKAMURA M, SADOSHIMA J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol 2018; 15:387-407
- NISHIMURA RA, OTTO CM, BONOW RO, et al. 2014 AHA/ACC Guideline for the management of patients with valvular heart disease. Circulation 2014; 129:e521-e643
- OPIE LH. Heart Physiology. From cell to circulation. 4th edition. Philadelphia: Lippincott Williams & Wilkins 2004, 402-430
- TOMANEK RJ. Response of the coronary vasculature to myocardial hypertrophy. J Am Coll Cardiol 1990; 15:528-33.
- WEBER KT, JALIL JE, JANICKI JS, et al. Myocardial collagen remodelling in pressure overload hypertrophy. Am J Hypertension 1989; 2:931-40.