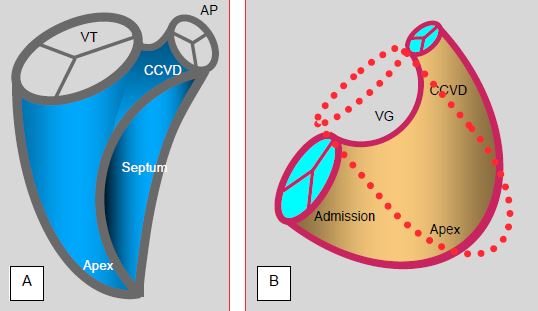
- Il est constitué d'une chambre d’admission basée sur l'anneau tricuspidien, d’un corps central très trabéculé en forme de croissant et d'une chambre de chasse (CCVD) cylindrique connectée à l’AP. Embryologiquement, le VD est issu de la partie antérieure du tube cardiaque (champ cardiaque secondaire) et la CCVD du champ cardiaque céphalique situé dans le mésoderme pharyngien (voir Annexe C, Figure C.13); cette dernière est dotée de davantage de récepteurs β que le reste du ventricule.
- Il est enroulé autour du VG ; sa section transversale est en demi-lune ; cette anatomie complique l’évaluation de sa fonction. Quel que soit le plan de coupe, la surface de la cavité du VD est plus petite que celle du VG (rapport SVD/SVG < 0.7) et l’apex du cœur est constitué par celui du VG (Figure 5.111).
- Il ne possède que deux couches musculaires : les fibres longitudinales sont sous-endocardiques et les fibres circulaires disposées à l’extérieur; ces dernières ne représentent que 25% de l'épaisseur de paroi. Une partie des fibres circulaires de la chambre d’admission et de l’apex est en continuité avec des fibres de la paroi du VG; ce dernier participe donc à la contraction circulaire du VD; a contrario, une défaillance gauche altère la performance du VD.
- Les sarcomères sont identiques dans les deux ventricules, mais la masse musculaire du VD est 1/6ème de celle du VG; l’épaisseur de la paroi libre du VD est de 4-5 mm environ.
- Le volume télédiastolique du VD est plus grand que celui du VG (50-100 mL/m2 versus 40-80 mL/m2); de ce fait, sa fraction d’éjection est plus basse (0.4 à 0.6) puisque les deux ventricules maintiennent le même volume systolique.
- Le VD est connecté à une valve tricuspide et possède trois muscles papillaires dont un est situé sur le septum. Sa surface interne est très fortement trabéculée et présente une travée musculaire qui traverse la cavité entre le septum et la paroi libre (bande modératrice); celle-ci est en continuité avec la crête supraventriculaire qui sépare la chambre d'admission de la CCVD par la bande septo-marginale. Sa chambre de chasse (CCVD), entièrement musculaire, présente une réponse inotrope plus forte que celle du reste du ventricule.
- La contraction du VD est liée à quatre mécanismes (voir Figure 5.34) :
- Contraction longitudinale base-apex-CCVD séquentielle en un mouvement péristaltique autour du VG; c’est l’élément principal (80% de l'éjaction).
- Raccourcissement vers l’intérieur de la paroi libre; le raccourcissement en court-axe est physiologiquement faible et peu performant.
- Rotation globale de 20-25°.
- Contribution importante du VG (environ 40-50% du volume d'éjection) par l’épaississement et la contraction longitudinale du septum interventricuaire, ainsi que par la traction sur les fibres circulaires à la jonction VG-VD.
- La contraction longitudinale du VD débute à la chambre d’admission (région sous-tricuspidienne) et se propage jusqu’à la CCVD comme un péristaltisme (délai 50-80 msec); le pic de pression est plus tardif que dans le VG. La phase de contraction isovolumétrique est quasi-inexistante, parce que la pression du VD atteint rapidement la valeur de la PAP qui est plus basse que celle de l’aorte.
- Le raccourcissement systolique longitiudinal représente 75% de la contraction; il est plus important que le raccourcissement en petit axe, qui peut être très modeste même dans les conditions physiologiques, et qui dépend davantage de la contraction des fibres septales du VG que de celles de la paroi libre du VD. La fraction d'éjection de la portion apicale est plus faible que celle de la chambre d'admission et du cône de chasse. Le flux intraventriculaire est relativement rectiligne, contrairement à celui du VG qui fait un large vortex.
- Le VG fournit 40-50% de la pression systolique du VD par la contraction du septum interventriculaire, qui assure une compression transverse et une contraction longitudinale de la cavité droite, et par la traction des fibres communes aux deux ventricules, qui contribuent à la contraction circulaire au niveau des sillons interventriculaires [23].
- Une obstruction dynamique de la chambre de chasse droite (effet CMO avec gradient jusqu’à 25 mmHg) peut survenir en cas d’instabilité hémodynamique accompagnée d’hypovolémie et de stimulation catécholaminergique parce que la CCVD est très richement dotée en récepteurs β [10].
- L’hypertrophie droite des cardiopathies congénitales s’accompagne d’une modification de la structure pariétale : le VD conserve une structure myocardique fœtale et développe trois couches de fibres au lieu de deux; il présente une structure qui ressemble à celle du VG.
- Situé antérieurement par rapport au VG, le VD court davantage de risque en cas de traumatisme thoracique par choc frontal.
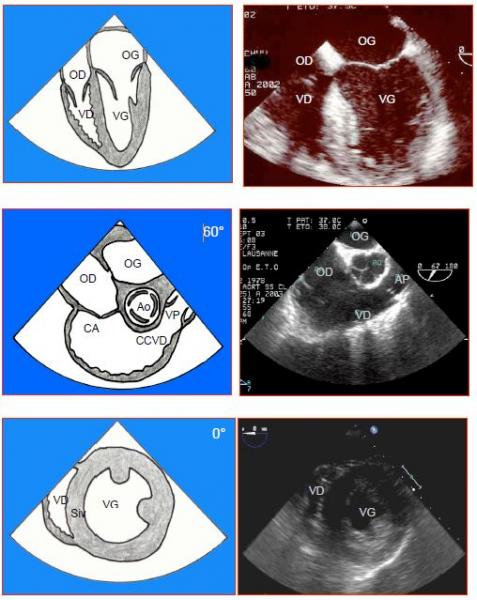
Figure 5.111 : Aspect échocardiographique transoesophagien du VD. A : vue 4-cavités (0°) ; l’apex est constitué par le VG, la surface du VD est de 0.6 fois celle du VG. B : vue à 60° de la chambre d’adminssion (CA) et de la chambre de chasse (CCVD) ; le VD est enroulé autour du VG. C : vue transgastrique (0°) ; la section du VD est en demi-lune. VP : valve pulmonaire. Siv : septum interventriculaire [2].
|
Différences structurelles entre les ventricules |
||
|
|
VG |
VD |
|
Forme |
conique en obus |
croissant enroulé autour du VG |
|
La contraction du VD comprend 4 mouvements différents,
- Contraction longitudinale péristaltique de la base vers l'apex et la CCVD, mouvement de propulsion principal
- Contraction radiaire de la paroi libre; contribution peu importante au volume systolique
- Torsion de 20-25°
- Apport important du VG: épaississement du septum et traction sur les fibres circulaires communes aux deux ventricules
(40-50% de l'éjection systolique) |
||
- PAP 20/10 - 30/15 mmHg (PAPmoy 10-19 mmHg);
- RAP : 65-160 dynes•s•cm-5 (1-2 U Wood);
- 1 U Wood = (PAPm – PAPO) / DC, en mmHg•min•L-1; on obtient un résultat en dynes•s•cm-5 en multipliant par 80.

Figure 5.112 : Diagramme Pression – Volume du ventricule droit. Comparée à celle du VG (voir Figure 5.52), la boucle Pression – Volume n’a pas une forme de quadrilatère mais plus ou moins de triangle. La pente Emax est plus faible et la courbe de compliance plus plate. 1 : point télédiastolique. 1 → 2 : contraction isovolumétrique (quasi-inexistante). 2 : début de l’éjection. 2 → 3 : phase de l’éjection systolique. 3 : point télésystolique. 3 → 4 : relaxation isovolumétrique (quasi-inexistante). 4 : début du remplissage. 4 → 1 : remplissage diastolique [19].
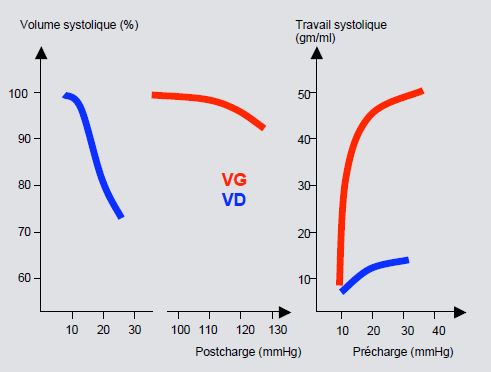
Figure 5.113 : Effets des variations de précharge et de postcharge sur les ventricules droit (en bleu) et gauche (en rouge) [19]. On voit l'extrême sensibilité du VD à l'élévation de sa postcharge, mais sa stabilité lors des variations de précharge.
- Le NO• est un vasodilatateur pulmonaire actif, puissant et rapide, synthétisé dans l’endothélium vasculaire en fonction de la pression et de la pulsatilité artérielles ; sa durée de vie est brève (10-60 sec). Il agit en augmentant l’activité du cGMP. Il inhibe l’agrégation plaquettaire [7].
- Les prostaglandines (PG) sont activement synthétisées dans les tissus vasculaires pulmonaires. La PGI1 et la PGE2 sont vasodilatatrices, alors que la PGF2α et la PGA2 sont vasoconstrictrices. Elles inhibent toutes l’agrégation plaquettaire [28].
- L’acidose, l’hypercapnie et l’hypoxie provoquent une vasoconstriction puissante à cause de l’augmentation locale de la concentration en ion H+. Le mécanisme est une augmentation de la [Ca2+] intracellulaire dans la musculature lisse des vaisseaux (voir Figure 5.127).
- L’endothéline (ET1), la sérotonine et l’angiotensine II sont des vasoconstricteurs pulmonaires. Ils stimulent également la croissance des fibroblastes, ainsi que l’hypertrophie et l’hyperplasie de la musculature lisse. Les facteurs qui règlent leur sécrétion sont le flux sanguin, la pulsatilité, le stress de la paroi vasculaire et l’hypoxie [6,22,25].
- Les récepteurs α1 sont actifs mais très peu nombreux dans l’arbre vasculaire pulmonaire ; leur existence même reste controversée. Aux dosages habituels, les stimulants α comme la nor-adrénaline, les anorexigènes et la cocaïne provoquent une vasoconstriction beaucoup plus faible que dans les vaisseaux systémiques. Ces substances n’agissent qu’à concentration élevée et/ou chronique [16]. La phentolamine induit une vasodilatation.
- Comme le rapport entre les récepteurs V1 (vasoconstriction) et V2 (vasodilatation) est en faveur des seconds dans le lit pulmonaire, la vasopressine a un effet vasodilatateur pulmonaire à faible dosage (< 0.07 UI/min), mais un effet vasoconstricteur seulement à forte dose (> 0.2 U/min) [9].
- L’innervation sympathique vasculaire pulmonaire est peu développée. Les récepteurs β1 provoquent une vasodilatation, mais leur activité n’est pas essentielle à son maintien. De ce fait, le bloc de la péridurale thoracique haute (C7-D4) tend plutôt à inhiber la vasodilatation pulmonaire active chez l’individu sain. En cas d’hypertension pulmonaire chronique, par contre, la population de récepteurs α1 est augmentée et le bloc sympathique peut contribuer à baisser les RAP [3,26].
- L'augmentation de sa postcharge: hypertension pulmonaire artérielle (précapillaire) et veineuse (postcapillaire);
- L'augmentation de sa précharge: surcharge de volume (cardiopathie congénitale, insuffisance tricuspidienne).
| Fonction du VD (I) |
|
Régime de pression du VD: 20/0 – 30/5 mmHg, RAP: 65 – 150 dynes•s•cm-5 (1-2 U Wood).
Boucle P/V triangulaire, l'éjection commençant pendant la contraction isovolumétrique.
Travail VD = 0.25 travail VG.
Courbe de Starling très aplatie ; le débit du VD est peu sensible aux variations de sa précharge mais très sensible à celles de sa postcharge (dilatation si augmentation aiguë).
Le VD est très sensible aux variations ventilatoires de la Pit (IPPV, PEEP).
L'arbre vasculaire pulmonaire est maintenu en vasodilatation de manière active (NO•, prostaglandines, faible innervation α, prédominance de récepteurs β). L'acidose, l'hypoxie, l'hypercapnie et l'hypothermie provoquent une vasoconstriction pulmonaire.
Le VD dilate en cas d'HTAP aiguë (pression maximale développée par un VD nornal: 50 mmHg, volume limite: 120 mL/m2). Si la postcharge est chroniquement élevée, le VD s'hypertrophie. Plus l'HVD est importante, plus le VD se comporte comme un VG et devient résistant à la postcharge mais sensible à la précharge.
|
Interdépendance ventriculaire
- Le VD assure la précharge du VG ; sa défaillance entraîne un défaut de remplissage gauche, donc un petit volume systolique et un bas débit systémique (temps de transit pulmonaire: 2-5 cycles cardiaques), qui à son tour compromettent la fonction et la perfusion coronarienne du VD.
- Les ventricules partagent une paroi commune, le septum interventriculaire (SIV), dont la position est définie par le gradient de pression entre le VD et le VG ; la contraction du SIV dépend essentiellement du VG et le fait normalement bomber dans le VD en systole.
- Le péricarde qui enveloppe les quatre chambres cardiaques représente une butée à l’augmentation de volume des ventricules ; la dilatation de l’un comprime l’autre. Plus leur volume diastolique est grand, plus le couplage entre eux est serré (il est de 1:1 lorsque la PtdVD est > 15 mmHg et la PtdVG > 20 mmHg) ; en hypovolémie, ce couplage diminue [20].
- La composante due à la contraction du VG représente jusqu’à 50% de la pression systolique générée dans le VD. Plus la surcharge de ce dernier augmente, plus l’assistance du VG est importante.
- Plus mince, le VD se dilate beaucoup plus facilement que le VG et empiète en diastole sur le volume du VG par un basculement du septum interventriculaire (effet Bernheim).
- La bascule du septum interventriculaire dans le VG en diastole en cas de dilatation du VD (défaillance et/ou surcharge de volume) ou en diastole et systole en cas de surcharge de pression du VD perturbe le remplissage et l’éjection du VG, dont le débit baisse. D’autre part, les modifications de la position du septum ventriculaire ampute le VD de l’aide fonctionnelle du VG à son éjection.
- En cas de surcharge de pression, la durée de la contraction systolique du VD s’allonge et se continue lorsque le VG est déjà en diastole, ce qui occasionne une bascule du septum dans ce dernier et une désynchronisation de sa contraction.
- La réduction de volume du VD s’il est dilaté (nitroglycérine, diurétique) améliore la performance du VG. L’augmentation de la postcharge du VG s’il est comprimé élève sa pression, repositionne le septum et améliore la performance du VD.
- La mise en route d’une assistance univentriculaire gauche peut non seulement démasquer une insuffisance droite sous-jacente mais encore supprimer le soutien du septum à l’éjection droite par décompression du VG.
- La perfusion coronarienne droite dépend du rapport de pression entre le VD et l’aorte en systole et en diastole.

Figure 5.114 : Défaillance ventriculaire droite. Bascule du septum inter-ventriculaire dans le VG lors de surcharge pulmonaire et/ou de défaillance ventriculaire droite. Le septum interauriculaire bombe dans l’OG. Le septum interventriculaire bombe dans le VG et en réduit considérablement le volume diastolique (effet Bernheim), comme en témoigne ici le rétrécissement de sa surface en diastole (la valve mitrale est ouverte). Le remplissage diastolique du VG est handicapé, et le volume systolique baisse. A: schématisation d’une vue 4-cavités. B : vue échocardiographique transoesophagienne 4-cavités (maladie thrombo-embolique pulmonaire chronique avec hypertension pulmonaire).

Figure 5.115 : Vues en court-axe des ventricules (échocardiographie transoesophagienne) en diastole et en systole lors de surcharge du VD. A : Image normale; le VD est enroulé en croissant autour du VG. B : Surcharge de volume. Le septum interventriculaire bombe dans le VG en diastole (effet Bernheim), mais non en systole car la pression et la force de contraction du VG restent supérieures à celles du VD dilaté. C : Surcharge de pression. Le septum interventriculaire bombe dans le VG aux deux temps car la pression du VD hypertrophié est capable de repousser le VG en télésystole. PAL : pilier antéro-latéral. PPM : pilier postéro-médian [Adapté de: Bettex DA, Chassot PG. Echocardiographie transoesophagienne en anesthesie-réanimation. Paris: Masson, Williams & Wilkins, 1997, Figures 7.22 et 7.23].
- Le flux coronarien droit est continu en diastole et en systole. (Figure 5.116).
- La réserve métabolique du VD est plus importante que celle du VG:
- La mVO2 droite (4.5 mL/min/100 g) est la moitié de la gauche (10 mL/min/100 g);
- L'extraction d'O2 est plus faible (40% au lieu de 70-80%); à l'effort, le VD augmente son extraction d'O2.
- La densité capillaire intramyocardique est plus importante, bien que le nombre de capillaires fonctionnels au repos soit plus faible.
- La région sous-endocardique n'est pas prétéritée comme à gauche parce que la compression transmurale est uniforme et faible.
- Le myocarde droit bénéficie d'une collatéralisation depuis la gauche (artère de la bande modératrice issue de l'IVA).
- La stimulation sympathique se traduit par une prédominance de réponse vasoconstrictrice (prédominance de récepteurs α); l'incidence de vasospasme (angor de Prinzmetal) est plus élevée; la nor-adrénaline peut diminuer le flux coronarien droit.
- La plage d'autorégulation du flux coronarien par rapport à la pression est faible; la baisse de pression de perfusion provoquée par une sténose de la CD n'est pas compensée par une vasodilatation du lit vasculaire distal à cause de la faible production de NO par l'endothélium.

Figure 5.116 : Flux coronarien droit et gauche. A: schéma du flux coronarien systolo-diastolique dans le ventricule droit (trait bleu) et presqu'exclusivement diastolique dans le ventricule gauche (trait rouge). B: enregistrement Doppler du flux dans l'artère interventriculaire antérieure (IVA) à l'échocardiographie transeosophagienne. C: enregistrement Doppler du flux dans la coronaire droite à l'échocardiographie transeosophagienne. Le flux est situé en dessous de la ligne de base parce qu’il s’éloigne du capteur situé dans l’œsophage.
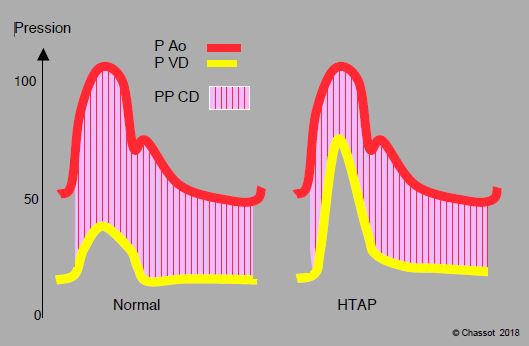
Figure 5.117 : Pression de perfusion coronarienne du VD. Vu la différence de pression entre l’aorte (P Ao) et le VD (P VD), la perfusion coronaire droite (PP CD) est systolo-diastolique. En cas d’hypertension pulmonaire (HTAP), la composante systolique de la perfusion droite est compromise. Le VD peut souffrir d’ischémie même si la perfusion diastolique est maintenue. Dans le schéma, la pression aortique est inchangée; dans la réalité, une simple hypotension systémique compromet la perfusion coronarienne droite en cas d'HTAP. Une perfusion de vasopresseur systémique est nécessaire pour maintenir l’adéquation de la composante systolique en cas d’HTAP. Pression en mmHg.
| Fonction du VD (II) |
|
L'interdépendance ventriculaire augmente en cas de dilatation droite et/ou gauche et en cas de restriction péricardique. Lors de surcharge de volume, le VD bombe dans le VG en diastole et restreint le remplissage du VG (effet Bernheim). Lors de surcharge de pression, le VD bombe dans le VG en diastole et en systole (allongement de la durée d'éjection droite, pression VD momentanément > pression VG et dyskinésie septale).
La perfusion coronarienne du VD est systolo-diastolique. Si la PsystVD est > PAmoy aortique, la perfusion coronarienne systolique baisse (ischémie VD); si le bombement du septum dans le VD diminue, l'aide du VG au VD devient inopérante. D'où l'importance des vasoconstricteurs systémiques dans la prise en charge de l'hypertension pulmonaire et de l'insuffisance droite, pour élever la pression du VG et repositionner le septum.
|
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2019
- ATHERTON JJ, MOORE TD, LELE SS, et al. Diastolic ventricular interaction in chronic heart failure. Lancet 1997; 349:1720-4
- BETTEX DA, CHASSOT PG. Echocardiographie transoesophagienne en anesthesia-réanimation. Paris: Masson, Williams & Wilkins, 1997, 245
- BLAISE G, LANGLEBEN D, HUBERT B. Pulmonary arterial hypertension. Anesthesiology 2003; 99:1415-32
- BRONICKI RA, BADEN HP. Pathophysiology of right ventricular failure in pulmonary hypertension. Pediatr Crit Care Med 2010; 11(Suppl):S15-S22
- BUCKBERG GD, HOFFMAN JIE. Right ventricular architecture responsible for mechanical performance: unifying role of ventricular septum. J Thorac Cardiovasc Surg 2014; 148:3166-71
- CACOUB P, DORENT R, NATAF P, et al. Endothelin-1 in the lungs of patients with pulmonary hypertension. Cardiovasc Res 1997; 33:196-203
- COOPER CJ, LANDZBERG MJ, ANDERSON TJ, et al. Role of the nitric oxide in the local regulation of pulmonary vascular resistance in humans. Circulation 1996; 93:266-71
- CRYSTAL GJ, PAGEL PS. Right ventricular perfusion. Physiology and clinical implications. Anesthesiology 2018; 128:202-18
- CURRIGAN DA, HUGHES RJ, WRIGHT CE, et al. Vasoconstrictor responses to vasopressor agents in human pulmonary and radial arteries: an in vitro study. Anesthesiology 2014; 121:930-6
- DENAULT AY, HADDAD F, JACOBSOHN E, et al. Perioperative right ventricular dysfunction. Curr Opin Anaesthesiol 2013; 26:71-81
- FENELEY MP, OLSEN CO, GLOWER DD, et al. Effect of acutely increased right ventricular afterload on work output of the left ventricle in conscious dog. Circ Res 1989; 65:135-145
- FISCHER LG, VAN AH, BURKLE H. Management of pulmonary hypertension: physiological and pharmacological considerations for anesthesiologists. Anesth Analg 2003; 96:1603-16
- FRIEDBERG MK, REDINGTON AN. Right versus left ventricular failure: differences, similarities, and interactions. Circulation 2014; 129:1033-44
- HADDAD F, COUTURE P, TOUSIGNANT C, DENAULT AY. The right ventricle in cardiac surgery, a perioperative perspective: I. Anatomy, physiology and assessment. Anesth Analg 2009; 108:407-21
- KONSTAM MA, UDELSON JE. Right heart failure. In: HOSENPUD JD, ed. Congestive heart failure. Pathophysiology, diagnosis and comprehensive approach to management. New York : Springer Verlag, 1994, 258-80
- KWAK YL, LEE CS, PARK YH, et al. The effect of phenylephrine and norepinephrine in patients with chronic pulmonary hypertension. Anaesthesia 2002; 57:9-14
- LA GERCHE A, BURNS AT, MOONEY DJ, et al. Exercise-induced right ventricular dysfunction and structural remodelling in endurance athletes. Eur Heart J 2012; 33:998-1006
- MARCUS JT, VONK NA, ROELEVEDL RJ, et al. Impaired left ventricular filling due to right ventricular pressure overload in primary pulmonary hypertension. Chest 2001; 119:1761-5
- McNEE W. Pathophysiology of cor pulmonale in chronic obstructive pulmonary disease. Part one. Am J Respir Crit Care Med 1994; 150:833-52
- MOORE TD, et al. Ventricular interaction and external constraint account for decreased stroke work during volume loading in CHF. Am J Physiol 2001; 281:H2385-H2391
- MORRIS-THURGOOD JA, FRENNEAUX MP. Diastolic ventricular interaction and ventricular diastolic filling. Heart Fail Rev 2000; 5:307-23
- RICH S, McLAUGHLIN VV. Pulmonary hypertension. In: ZIPES DP, et al, eds. Braunwald’s heart disease. A textbook of cardiovascular medicine. Philadelphia: Elsevier Saunders, 2005, 1807-42
- SANTAMORE WP, GRAY L. Significant left ventricular contributions to right ventricular function. Mechanisms and clinical implications. Chest 1995; 107:1134-45
- SANZ J, SANCHEZ-QUINTANA D, BOSSONE E, et al. Anatomy, function, and dysfunction of the right ventricle. JACC state-of-the-art review. J Am Coll Cardiol 2019; 73:1462-82
- SCHUSTER DP, CROUCH EC, PARKS WC, et al. Angiotensin converting enzyme expression in primary pulmonary hypertension, Am J Respir Crit Care Med 1996; 154:1087
- SUBRAMANIAM K, YARED JP. Management of pulmonary hypertension in the operating room. Semin Cardiothor Vasc Anesth 2007; 11:119-36
- VANDENHEUVEL MA, BOUCHEZ S, WOUTERS P, DE HERT SG. A pathophysiological approach towards right ventricular function and failure. Eur J Anaesthesiol 2013; 30:386-94
- VANE JR, ANGGARD EE, BOTTING RM. Regulatory functions in the vascular endothelium. N Engl J Med 1990; 323:27-34
- VENTETUOLO CE, KLINGER JR. Management of acute right ventricular failure in the intensive care unit. Ann Am Thorac Soc 2014; 11:811-22
- VONK-NOORDEGRAAF A, HADDAD F, CHIN KM, et al. Right heart adaptation to pulmonary arterial hypertension. J Am Coll Cardiol 2013; 62:D22-33
- WARNES CA. Adult congenital heart disease. Importance of the right ventricle. J Am Coll Cardiol 2009; 54:1903-10
- ZILE MR, BRUTSAERT DL. New concept in diastolic dysfunction and diastolic heart failure: Part I. Diagnosis, prognosis, and measurement of diastolic function. Circulation 2002; 105:1387-93