La membrane des myocytes présente de longues et fines invaginations, les tubules T, qui amènent le liquide extracellulaire au voisinage des organelles intracellulaires. Les récepteurs membranaires, qui sont situés sur ces tubules T, se trouvent donc tout proches du réticulum sarcoplasmique (RS) et des mitochondries. Deux voies sont possibles pour déclencher une contraction myocardique et gérer son intensité : les récepteurs β et les canaux calciques L.
Voie des récepteurs β
Le récepteur β est un complexe protéique de sept spires à cheval sur la membrane cellulaire (Figure 5.1). Il est très stéréospécifique : il faut une configuration moléculaire précise pour le stimuler (la meilleure spécificité est celle de l’isoprénaline). Il possède aussi une haute affinité : de faibles concentrations circulantes de catécholamines (10-9-10-10 mol/L) suffisent à déclencher la stimulation. Il comprend des zones fonctionnant comme agonistes et des zones dont la stimulation induit un effet antagoniste. Il est couplé à deux protéines G intracellulaires, l’une stimulatrice (Gs) et l’autre inhibitrice (Gi) (il existe plusieurs types de protéines G dont les principaux sont les types Gs, Gi et Gq). Il possède également des points dont la phosphorylation induit sa désensibilisation par découplage de la protéine Gs [7,10].
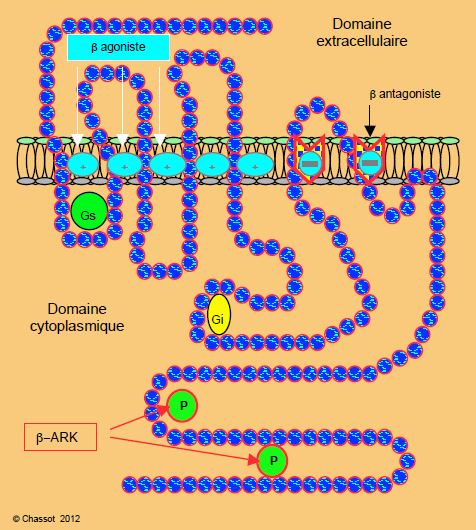
Figure 5.1 : Configuration d’un récepteur β. Il présente une structure en sept spires à cheval sur la membrane cellulaire. La stimulation de certaines zones a un effet agoniste, alors que celle d’autres zones a un effet antagoniste. Gs : protéine G stimulatrice. Gi : protéine G inhibitrice. P : points de phosphorylation pour la β-ARK (β-Agonist-Receptor-Kinase) qui désactive le récepteur.
Dans les ventricules, le 80% des récepteurs est de type β1, à effet essentiellement inotrope positif, alors que dans les oreillettes la proportion est de 60% β1 et 40% β2 (respectivement inotrope et chronotrope) [4,5]. Ainsi une catécholamine à effet β1 pur comme la dobutamine a un effet essentiellement inotrope, alors que les catécholamines à effet β2 provoquent en plus une tachycardie. La plupart des récepteurs β non-cardiaques sont de type β2 ; leur stimulation cause une vasodilatation artérielle et une accélération métabolique. Dans un cœur normal, les récepteurs β2 et α1 représentent environ 10% chacun de la population totale. Il existe encore des récepteurs β3 (1%) qui ne sont actifs que lors de fortes stimulations sympathiques ou dans l’insuffisance ventriculaire ; ils ont un effet inotrope négatif et servent à limiter l’effet cardiostimulant des β1 et β2 [3,8].
Lorsqu’il est stimulé, le récepteur β1 active la protéine Gs qui stimule un enzyme, l’adénylate-cyclase, seul capable de produire de l'AMPc (second messenger) à partir de l’adénosine-triphosphate (ATP) (Figure 5.2) [7]. L'AMPc, produit en concentration minime (10-10 mol/L), active la protéine-kinase A (PK-A) qui va induire la phosphorylation de plusieurs protéines et enzymes intracellulaires, dont les récepteurs ryanodine (R-Ry) situés sur le réticulum sarcoplasmique (RS) ; ces derniers, une fois activés, permettent la libération de Ca2+ à partir du RS [2]. Les R-RY sont inhibés par le tacrolimus et le sirolimus utilisés dans les stents actifs.
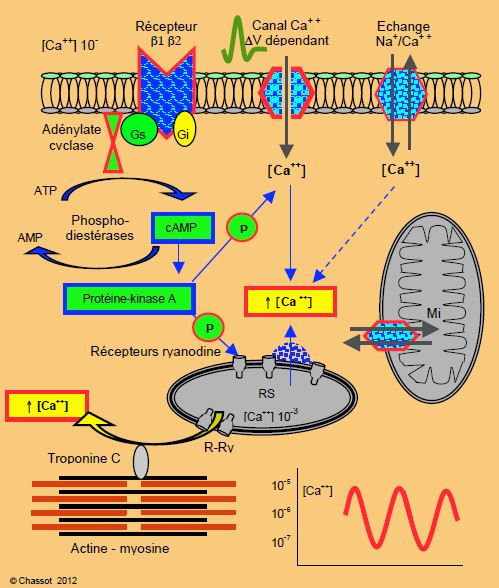
Figure 5.2 : Schématisation du couplage excitation - contraction myocardique par la voie des récepteurs β et par celle des canaux calciques L dépendant de la dépolarisation systolique (ΔV de l’onde QRS) (description détaillée dans le texte). Gs : protéine G stimulatrice. Gi : protéine G inhibitrice. cAMP : adénosine monophosphate cyclique. ATP : adénosine triphosphate. RS : réticulum sarcoplasmique. R-Ry : récepteurs ryanodine.
Le taux d'AMPc est maintenu très bas par l’activité des phospho-diestérases-3 (PDE-3), qui sont responsables de la dégradation de l'AMPc en AMP inactif. Leur inhibition par les anti-PDE-3 (amrinone, milrinone) maintient un taux élevé d'AMPc par un circuit indépendant des récepteurs β, et provoque de ce fait un effet inotrope positif non-catécholaminergique.
Voie des canaux calciques L
La dépolarisation électrique de la membrane cellulaire par le potentiel d’action ouvre les canaux calciques L, ou canaux voltage-dépendents, pendant 0.2 msec [9]. Une petite quantité de Ca2+ pénètre alors dans la cellule, et a pour effet de stimuler une libération de Ca2+ par le RS qui est 100 fois plus importante (calcium-induced calcium release) ; cette libération se fait par les récepteurs ryanodine (R-RY). Ce système amplificateur, qui prend 40-100 msec à une fréquence cardiaque de 60/min, permet d’obtenir une élévation importante de la concentration de Ca2+ libre dans le cytoplasme sans pour autant requérir un important passage de Ca2+ à travers la membrane cellulaire [2].
Variations de la [Ca2+]
Seule une fraction minime du Ca2+ entre et sort du cardiomyocyte lors de la contraction; la majeure partie (> 90%) ne fait que des allers-retours entre le cytoplasme et le RS. Dans ce dernier, la [Ca2+] est la plus élevée de la cellule: 10-3 M; elle est voisine de celle qui règne à l’extérieur de la membrane cellulaire [1]. Au repos, la [Ca2+]i est très basse: 10-7 M; elle s'élève à 10-5 M pendant 50-200 msec en systole. L’intensité de la contraction myocardique est fonction directe de l’amplitude de la variation du taux de Ca2+ libre dans le sarcoplasme.
L’ajustement global de la quantité de Ca2+ présent dans la cellule se fait par deux voies.
- Les canaux calciques L qui laissent entrer le Ca2+ lors du potentiel d’action;
- Les canaux Na+-Ca2+ qui échangent le Ca2+ et le Na+ et rejettent le premier à l’extérieur pendant la diastole.
Ces canaux ne sont responsables que du 7-8% du trafic calcique total. La capacité des mitochondries à repomper le Ca2+ est très limitée, car une concentration trop élevée de Ca2+ y freine la production d’ATP ; la [Ca2+]m y est voisine de celle du cytoplasme.
Le calcium ionisé libéré par la stimulation se fixe sur la troponine-C (TnC), ce qui modifie la configuration de la tropomyosine liée à l’actine et lève l'inhibition mécanique que cette dernière exerce sur le complexe actine-myosine ; la contraction est déclenchée (voir Contraction myocardique, Physiologie). Une stimulation β augmente ce processus : c’est l’effet inotrope positif.
La stimulation β a aussi la propriété de faciliter la phosphorylation de la troponine I (TnI inhibitrice) par la PK-A et la PK-C générées par l'AMPc ; la TnI abaisse l’affinité de la TnC pour le Ca2+, ce qui facilite leur dissociation à la fin de la systole. Cette amélioration de la relaxation diastolique correspond à l’effet lusitrope positif des amines [6]. L'énergie consommée pendant la contraction est fournie par hydrolyse de l'ATP au moyen d'une ATPase qui fait partie de la myosine et dont l'activité est elle aussi réglée par le taux de Ca2+.
| Déclenchement de la contraction |
|
Deux voies: récepteurs β (catécholamines) et canaux calciques L (dépolarisation électrique).
Récepteurs intraventriculaires (cœur normal): 80% de récepteurs β1 (effet inotrope positif), 10% de récepteurs β2 (effet chronotrope) et 10% de récepteurs α1 (inotrope positif).
La stimulation β et/ou électrique provoque l'activation de l'AMPc et une libération de Ca2+ à partir du réticulum sarcoplasmique. Lorsque la [Ca2+] passe de 10-7 M à 10-5 M dans le cytoplasme, la contraction musculaire est déclenchée.
|
© CHASSOT PG Août 2010, dernière mise à jour Novembre 2018
Références
- BERS DM. Excitation-contraction coupling and cardiac contractile force. Boston: Kluwer Academic Publishers, 2001
- BERS DM. Cardiac excitation-contraction coupling. Nature 2002; 415:198-205
- BRODDE OE, DAUL A, MICHEL-REHNER L, et al. Agonist-induced desensitization of beta-adrenoreceptor function in humans. Circulation 1990; 81:914-21
- DEL MONTE F, KAUFMANN AJ, POOLE-WILSON PA, et al. Coexistence of functioning 1- and 2-adrenoreceptors in single myocytes from human ventricle. Circulation 1993; 88:854-63
- KOBILKA B. Molecular and cellular biology of adrenergic receptors. Trends Cardiovasc Med 1991; 1:189-94
- MORANO I. Tuning smooth muscle contraction by molecular motors. J Mol Med 2003; 81:481-7
- OPIE LH. Heart Physiology. From cell to circulation. Philadelphia: Lippincott-Williams & Wilkins, 2004, 648 pp
- POST SR, HAMMOND HK, INSEL PA. -adrenergic receptors and receptor signalling in heart failure. Annu Rev Pharmacol Toxicol 1999; 39:343-60
- WIER WG, EGAN TM, LOPEZ-LOPEZ JR, et al. Local control of excitation-contraction coupling in rat heart cells. J Physiol 1994; 474:463-71
- ZAUGG M, SCHAUB MC. Cellular mechanisms in sympatho-modulation of the heart. Br J Anaesth 2004; 93:34-52