Physiopathologie
L'hypertension pulmonaire (HTP) est définie par une PAP moy > 25 mmHg au repos ou des résistances vasculaires pulmonaires (RAP) > 300 dynes•cm•s-5 (voir Chapitre 12 Physiopathologie de l'HTP) [17,18]. L’hypertension artérielle pulmonaire (HTAP), rencontrée chez 5-10% des congénitaux, est plus spécifiquement une augmentation de la pression précapillaire. L'HTP est classée en 5 catégories [17,23,37].
- 1 - Hypertension artérielle pulmonaire (HTAP): HTAP des cardiopathies congénitales, HTAP primaire idiopathique, HTAP secondaire à des médicaments, à des infections, à l'âge.
- 1' - Maladie pulmonaire veino-occlusive.
- 1’’ - HTAP persistante du nouveau-né (2 :1'000 bébés).
- 2 - Hypertension pulmonaire (HTP) postcapillaire (PtdVG > 18 mmHg, PAPO > 15 mmHg, mais RAP < 3 U Wood et gradient transpulmonaire < 12 mmHg): défaillance systolique ou insuffisance diastolique restrictive du VG, valvulopathie mitrale.
- 3 - HTAP associée à l’hypoxie alvéolaire: BPCO, emphysème, SDRA, apnée du sommeil, hypoxie d’altitude.
- 4 - HTAP liée à la maladie thrombo-embolique pulmonaire chronique.
- 5 - HTAP d’origine multifactorielle non éclaircie.
Dans les groupes 1, 3, 4 et 5, l’HTAP est précapillaire; la POG et la PAPO sont normales (PAPO < 15 mmHg), et le gradient transpulmonaire (GTP = PAPm – POG, ou PAPm - PAPO) est > 12 mmHg; les RAP sont élevées (> 240 dynes•s•cm-5 ou > 3 U Wood). L’HTP liée aux pathologies du cœur gauche (groupe 2) est primairement postcapillaire: POG et PAPO élevées (> 15 mmHg), RAP et gradient transpulmonaire bas (GTP < 12 mmHg). Si l’on extrait la pression artérielle pulmonaire de l'équation qui calcule les résistances artérielles, on trouve : PAP ≈ POG + (DC • RAP). La pression de l’AP dépend donc de trois facteurs: la pression de l’OG (stase gauche, HTP postcapillaire), le débit pulmonaire (shunt gauche-droit, HTP hyperkinétique) et les résistances artérielles pulmonaires (hypertension artérielle pulmonaire, HTAP précapillaire) (Figure 15.65) [27,31].
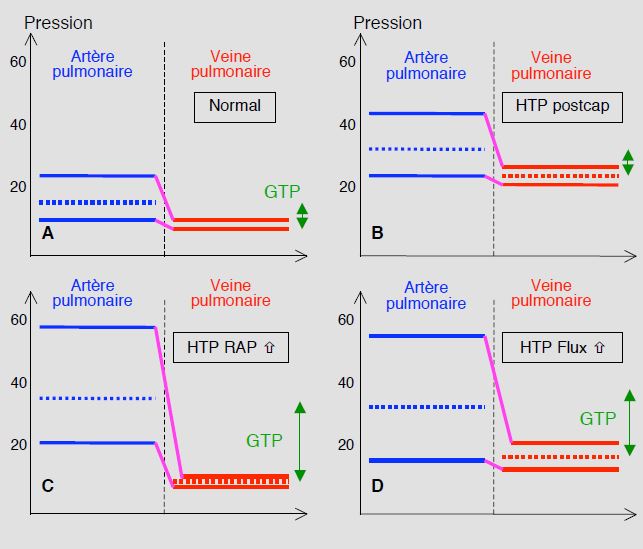
Figure 15.65: Classification schématique des différents mécanismes de l'hypertension pulmonaire (HTP) [31]. A: situation normale; le gradient transpulmonaire (GTP, double flèche verte) est < 12 mmHg. B: HTP postcapillaire ou passive; la pression veineuse est élevée, ce qui entraine une élévation secondaire de la PAP mais le gradient transpulmonaire reste normal. C: HTP précapillaire ou résistive; le mécanisme primaire est une élévation des RAP; la pression veineuse est normale et le GTP est très élevé (> 12 mmHg). D: HTP hyperkinétique secondaire à une augmentation du flux sanguin pulmonaire (shunt G-D); la PAP est élevée, mais la pression veineuse est légèrement supérieure à la norme, ce qui réduit un peu l'augmentation du GTP. Comme les RAP augmentent secondairement à la surcharge de flux, l'HTP des congénitaux adultes est un mélange de C et de D.
Chez les congénitaux, la cause la plus fréquente d'HTAP est un shunt G-D non restrictif. Elle survient dans 50% des cas de CIV et de canal AV, où il y a surcharge de volume et de pression, mais dans seulement 10% des CIA, où il y a surcharge de volume uniquement. Elle apparaît dans l'enfance déjà lors de CIV, mais seulement à l'âge adulte en cas de CIA [30]. Le stress pariétal vasculaire provoqué par le flux pulmonaire excessif cause une extension de la musculature lisse dans les vaisseaux périphériques qui ne sont normalement pas musculaires; à ce stade, la pression reste à la limite supérieure de la norme [34]. L'accroissement continu de pression est associé à une hypertrophie de la média des artères plus proximales, accompagnée d’une prolifération intimale progressive et d’une fibrose, puis d’épaississement de l’adventice, de réaction inflammatoire et de thrombus disséminés [32] (Figure 15.66).

Figure 15.66 : Image anatomo-pathologique d’une artériole pulmonaire en cas de syndrome d’Eisenmenger. 1 : hypertrophie de la média. 2 : prolifération intimale. 3 : fibrose [32]. La paroi d’une telle artère ne peut plus être collabée par les pressions ventilatoires en ventilation mécanique.
Finalement, le nombre des vaisseaux distaux diminue, l'HTAP est fixée et irréversible. C'est le syndrome d'Eisenmenger, caractérisé par une PAP moyenne > 50 mmHg au repos, des RAP > 800 dynes•cm•s-5 et un shunt bidirectionnel (shunt G-D + composante D-G) accompagné de cyanose [17]. Le renversement du shunt G-D est la principale origine de l'apparition soudaine d'une cyanose chez les congénitaux adultes [13]. L’irréversibilité de l’HTAP est démontrée par un test de vasoréactivité au NO, dans lequel la réversibilité potentielle est définie par une réduction de la PAP moyenne de > 10 mmHg et une baisse de celle-ci à ≤ 40 mmHg, sans diminution du débit cardiaque [5,18].
Les deux ventricules ont une masse égale pendant la vie intra-utérine ; s’il est exposé dès la naissance à une postcharge élevée, le VD conserve une structure fœtale et s’hypertrophie parallèlement au VG [8]. Il résiste mieux à l'accroissement de postcharge que lorsque celle-ci survient secondairement pendant la vie adulte. Bien que la correction chirurgicale ne soit plus possible lorsque le rapport RAP/RAS est supérieur à 0.7, le devenir des congénitaux adultes est meilleur que celui des patients souffrant d'HTAP primaire, médicamenteuse ou thrombo-embolique [6,22,24].
- CIA: survie de 90% à 40 ans;
- Syndrome d'Eisenmenger: survie de 80% à 10 ans;
- HTAP primaire idiopathique: survie de 66% à 5 ans;
- Maladie thrombo-embolique: survie de 35% à 5 ans.
Le pronostic de l’HTAP est étroitement lié à la fonction droite, bien plus qu'à la valeur de la PAP. Il est sombre dès que le VD dysfonctionne et se dilate. En effet, l’HTAP est fonction de la capacité du VD à générer chroniquement des pressions pulmonaires élevées. Dès que le VD défaille, la PAP tend à redescendre, alors que la situation hémodynamique empire et que les RAS continuent à augmenter (Figure 15.67) [21].
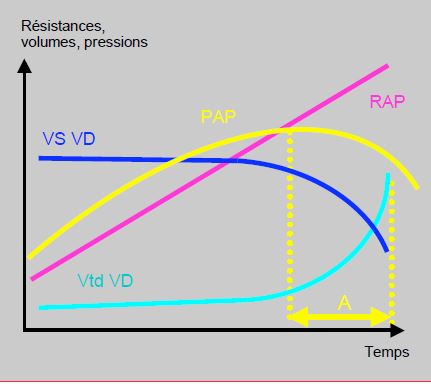
Figure 15.67 : Couplage entre le VD et la circulation pulmonaire. Schématisation de la relation entre les résistances artérielles pulmonaires (RAP), le volume systolique du VD (VS VD), le volume télédiastolique du VD (VtdVD) et la pression artérielle pulmonaire (PAP). L’HTAP est fonction de la capacité du VD à générer chroniquement des pressions pulmonaires élevées. Dès que le VD défaille, la PAP tend à redescendre, alors que la situation hémodynamique empire et que les RAS continuent à augmenter. Dans le laps de temps A (en jaune), la PAP mesurée est plus basse que précédemment, non par amélioration de la situation mais pas péjoration de fonction du VD. La gravité clinique et le pronostic de la maladie tiennent donc davantage à la fonction ventriculaire droite qu’à la valeur de la PAP en elle-même [21].
Pour l’anesthésiste, l'hypertension pulmonaire des congénitaux adultes présente certaines caractéristiques physio-pathologiques qui sont communes à toutes les formes d’HTAP, à l’exception de la première qui lui est propre [4,7,11,15,18,27].
- Une baisse de pression systémique par hypovolémie ou par vasodilatation systémique aggrave la composante D-G du shunt, diminue le débit pulmonaire et péjore la cyanose.
- Le débit pulmonaire est abaissé et fixe; toute compliance aux variations de débit et de volume circulant est perdue. Une augmentation du Qp est impossible lors d’une augmentation de la demande en O2: le malade est désaturé au moindre effort.
- L’hypotension systémique diminue la pression de perfusion coronaire, accentue la bascule du septum interventriculaire vers la gauche et menace la perfusion sous-endocardique du VD.
- Plus il est hypertrophié, plus le VD se comporte comme le VG; sa courbe de Starling se redresse et son débit devient dépendant de sa précharge. Dans ce cas, l’hypovolémie conduit à une baisse du débit pulmonaire et à une hypoxémie.
- En diastole, la pression du VD hypertrophié et surchargé est supérieure à celle du VG; le septum interventriculaire bombe dans le VG (effet Bernheim) et en réduit le remplissage diastolique. L’élévation de la postcharge gauche (vasoconstriction artérielle systémique) tend à replacer le septum dans sa position physiologique.
- Malgré l’épaississement des parois vasculaires, la réactivité des vaisseaux pulmonaires est conservée: l'hypercarbie, l'acidose et l'hypoxémie augmentent encore les RAP, alors que l'hyperventilation les abaisse [7,9]. L'anesthésiste doit donc éviter toute les manoeuvres qui peuvent aggraver les RAP [11]:
- Hypoventilation: hypercarbie, hypoxémie, atélectasies;
- Surpression endothoracique;
- Acidose;
- Hypothermie;
- Anémie (Hb < 100 g/L);
- Stimulation sympathique, stress, douleur.
L’HTAP est un des principaux déterminants du risque opératoire chez les congénitaux. Seules des interventions vitales sont envisageables chez ces patients [10].
Traitement de l’hypertension pulmonaire
L'anesthésiste doit prendre toutes les mesures possibles pour abaisser la pression pulmonaire (voir Chapitre 12 Traitement de l’HTAP). Il est capital que le patient soit profondément anesthésié (stress-free anaesthesia, fentanyl, sufentanil ou remifentanil en doses importantes) et qu'il reste normotherme. La première étape du traitement aigu de l’HTAP est ventilatoire, parce que l’alcalose respiratoire, l’inhalation de gaz et la nébulisation de substances sont les seules techniques qui permettent de baisser la pression artérielle pulmonaire sans hypotension systémique. L’inhalation présente le double avantage de ne pas causer d’hypotension ni de freiner la vasoconstriction pulmonaire hypoxique des zones non-ventilées, puisque les substances ne sont distribuées que dans les alvéoles ventilées ; de ce fait, l’hypoxémie ne s’aggrave pas (Tableau 15.6) [7,11,17,25, 27,38].

- Hyperventilation: l'hypocapnie (PaCO2 30-35 mm Hg) et l'alcalose (pH 7.5) ont un effet vasodilatateur pulmonaire sans effet sur le lit systémique. La FiO2 doit être élevée, les pressions de ventilations basses et le volume courant proche de la CRF (6-8 ml/kg.); c'est le pH et non la pCO2 qui règle le tonus vasculaire pulmonaire. La réactivité des RAP à l'acidose et à l'alcalose est accentuée en cas d'hypoxie et d'hypertension pulmonaire [7]. Le meilleur moyen de régler le ventilateur est d'assurer la pression de ventilation moyenne minimale (6-10 mmHg) pour une PetCO2 située entre 25 et 30 mmHg.
- Administration d’O2 (lunettes, masque) en dehors de la salle d’opération, bénéfique surtout lorsque l’érythrocytose est marquée (Ht > 55%).
Viennent ensuite les moyens pharmacologiques. Certaines substances peuvent être nébulisées dans le circuit respiratoire, et sont donc sans effet systémique [38,39].
- NO: 10 – 30 ppm dans le circuit inspiratoire du ventilateur; sans effet systémique, le NO est immédiatement inactivé par sa liaison avec l’hémoglobine, mais il altère la fonction plaquettaire. Dans les cas d'hypertension pulmonaire, ou dans les situations où la PAP doit rester basse (Fontan, par exemple), le NO doit être administré dès l’induction et/ou la reprise de la ventilation après CEC, avant que ne survienne une poussée hypertensive. Son sevrage peut être difficile à cause d’un effet rebond sur l’HTAP, qui peut être tempéré par l’administration de sildénafil, de prostacycline ou de milrinone. Si les RAP ne diminuent pas dans les 30 minutes qui suivent l’administration de NO•, la substance est inefficace et le traitement doit être interrompu [3].
- Prostacyclines : iloprost en spray nasal (Ilomedin®, 10-20 mcg en 20 min) à répéter toutes les 3-4 heures (demi-vie 30 minutes), ou en nébulisation continue (0.2-0.3 mL/min d’une solution de 10-20 mcg/mL). En traitement aigu, les prostacyclines ont un effet additif avec le NO•. Les prostacyclines diminuent l’agrégabilité plaquettaire.
- Inhibiteurs des phosphodiestérases-3 : nébulisée au lieu d’être administrée en perfusion, la milrinone a moins d’effet hypotenseur systémique et davantage d’efficacité pulmonaire. Nébulisation d’une solution de 1 mg/mL à 0.2-0.3 mL/min pendant 10-20 minutes, soit 50-80 mcg/kg en 10 min. Elle est moins coûteuse que le NO et les prostaglandines et ne provoque pas d’effet rebond lors de son interruption. D'autre part, l’iloprost et la milrinone sont plus faciles d’emploi que le NO•.
- Nitroglycérine : nébulisation à raison de 2.5 mcg/kg/min ; moins efficace que les précédents.
Il existe une série de substances susceptibles de baisser la PAP de manière aiguë par voie intraveineuse, mais tous ces vasodilatateurs pulmonaires présentent des effets hypotenseurs systémiques plus ou moins accentués [25,27].
- Prostaglandines: par stimulation de l'adénylate cyclase, elle accroissent le taux de cAMP et provoquent une vasodilatation; utilisées en traitement chronique et aigu, elles ont un effet additif avec le NO [35,36].
- Epoprosténol (Flolan®) en perfusion (2-5 ng/kg/min); demi-vie très brève (4-6 min);
- Iloprost (Ilomedin®) 10-20 mcg en 20 min; durée de l'effet: 60 minutes;
- Treprostinil (Remodulin®) s-cut ou iv (1.25-2.5 ng/kg/min); demi-vie 3 heures.
- Inhibiteur des phosphodiestérases-5: sildénafil (Revatio®, 3 x 10 mg/j iv); il s'utilise simultanément au NO et aux prostaglandines. Particulièrement efficace en association à la L-arginine [19,29]. Les IPDE-5 sont la seule classe de vasodilatateurs pulmonaires ayant un effet bénéfique sur l’HTP liée aux pathologies du cœur gauche [20].
- Magnésium : le Mg2+ (5-10 mmol) a un certain effet dilatateur pulmonaire de type anticalcique.
- Arginine : par sa transformation en citrulline, le chlorure d’arginine (15 mg/kg/min) fournit le substrat nécessaire à la production de NO; elle est probablement indiquée après une CEC, parce que celle-ci abaisse le taux d’arginine.
- Citrulline : ce précurseur du NO• peut réduire l’HTAP des cardiopathies congénitales chez l’enfant (perfusion 9 mg/kg/heure); données pour l’instant limitées.
- Nitroglycérine (0.5-5 mcg/kg/min): léger effet vasodilatateur pulmonaire en sus de la diminution du volume télédiastolique du VD et de l'amélioration de l'insuffisance droite congestive à PVC haute. Risque d'hypotension systémique par baisse de précharge. Elle n'est indiquée chez les congénitaux qu’en cas d’insuffisance congestive du VD (insuffisance tricuspidienne ou pulmonaire massive).
- Nesiritide (charge 2 mcg/kg, perfusion 0.01-0.03 mcg/kg/min): ce dérivé du BNP abaisse les RAP et améliore l’hémodynamique en cas d’insuffisance droite sur HTAP. Son impact clinique est probablement minime.
- Bloqueurs calciques: contre-indiqués chez les congénitaux, car la vasodilatation systémique plus importante que la vasodilatation pulmonaire peut accentuer l’effet cyanogène d’un shunt bidirectionnel [18].
- Le maintien de la PAM systémique au-dessus de la PAP (PAM/PAPm > 3) avec un vasoconstricteur artériel (noradrénaline ou vasopressine) est essentiel pour trois raisons.
- Diminuer la composante D-G du shunt s'il y a lieu.
- Rétablir la position du septum interventriculaire (convexe dans le VD) et l’aide à l’éjection droite réalisée par la contraction du VG ; un augmentation de postcharge gauche est nécessaire pour faire bomber le septum dans le VD en systole.
- Maintenir la perfusion coronarienne du VD, car celui-ci ne peut plus compter que sur la composante systolique du flux coronaire si la PAP se rapproche de la PAM [38].
La performance du VD est capitale pour assurer l'équilibre hémodynamique. En cas de dysfonction droite, plusieurs agents inotropes positifs ont une activité vasodilatatrice pulmonaire en plus de leur effet cardiostimulant et sont donc des choix préférentiels dans le cadre de l'hypertension pulmonaire.
- Dobutamine (Dobutrex®): par augmentation de la cAMP (stimulation β1), elle induit une vasodilatation pulmonaire significative.
- Inodilatateurs: les anti-phosphodiestérases-3 diminuent la PAP et les RAP, et augmentent la FE par effet inotrope positif; amrinone (Inocor®), milrinone (Corotrop® 0.5 mcg/kg/min).
- La combinaison adrénaline + milrinone est en général la plus efficace parce qu’elle équilibre l’effet inotrope, vasodilatateur pulmonaire et vasoconstricteur systémique.
- Levosimendan (dose de charge 6 mcg/kg, perfusion 0.05-0.2 mcg/kg/min): il diminue la PAP (postcharge droite) et augmente la contractilité ventriculaire par sensibilisation de la troponine au Ca2+. Il est le seul agent inotrope qui baisse la mortalité [33].
- Isoprénaline (Isuprel®): très efficace, mais provoque une tachycardie et une hypotension systémique importantes (stimulation β1 + β2).
- En peropératoire, ouverture ou non-fermeture du péricarde et du sternum pour décomprimer le VD et diminuer l'interdépendance ventriculaire.
- Assistance ventriculaire: ECMO, dispositif d'assistance du VD (Impella™, etc).
L'équilibre hémodynamique des congénitaux souffrant d'HTAP est très précaire. Il est donc capital d'avoir une attitude proactive et de mettre en route un traitement hypotenseur dès que la PAP commence à s'élever (PAPm > 0.5 PAM). Une élévation de la PVC et/ou une baisse du volume systolique sont des signaux d'alerte pour soutenir pharmacologiquement la fonction droite. De même, la surveillance du VD à l'ETO permet de démarrer un support inotrope dès que sa performance diminue ou que son volume augmente.
Les patients qui souffrent d'HTAP sont le plus souvent sous traitement chronique de vasodilatateur pulmonaire. Ce traitement doit être scrupuleusement maintenu en périopératoire [17,18].
- Inhibiteurs de l’endothéline: Bosentan (Tracleer®, 125-250 mg 2 x/j), sitaxsentan (100 mg/j);
- Inhibiteurs des phosphodiestérases-5: sildénafil (Viagra®, Revatio®, 3 x 20 mg/j po), tadalafil, vardenafil;
- Prostaglandines: beraprost, iloprost;
- Stimulant du cGMP: riociguat (Adempas®);
- Losartan (anti-angiotensine) à raison de 50-100 mg/j.
- Les bloqueurs calciques sont inutiles, voir dangereux, chez les congénitaux.
Les vaisseaux pulmonaires étant quasi dépourvus de récepteurs α1, les vasodilatateurs artériolaires (nitroprussiate, phentolamine) ont essentiellement un effet systémique et ne sont pas utiles dans le traitement sélectif de l'HTAP. Ils ont un effet catastrophique en présence de shunt D-G ou d'alimentation du débit pulmonaire par une vicariance systémique (shunt G-D de Blalock, collatérales aorto-pulmonaires). Vu cette faible population en récepteurs α, la nor-adrénaline et la néosynéphrine ont un effet vasoconstricteur pulmonaire négligeable. La noradrénaline augmente le rapport RAP/RAS, alors que la vasopressine le diminue, car l’effet vasoconstricteur systémique de cette dernière ne s’accompagne pas d’une hausse des RAP vu la prédominance des récepteurs V2 vasodilatateurs sur les récepteurs V1 vasoconstricteurs dans le lit pulmonaire [26]. Les vasodilatateurs pulmonaires sont contre-indiqués dans l'HTP secondaire à des maladies du cœur gauche (type 2), car ils augmentent le retour veineux à l'OG et font courir un risque de surcharge et d'OAP; seuls les IPDE-5 ont une place dans cette situation, pour autant que le gradient transpulmonaire (PAPm – PAPO) soit > 12 mmHg.
Dans les cas de surcharge droite extrême, une septostomie auriculaire (CIA iatrogène) permet de décomprimer le VD et d’améliorer la précharge du VG, mais au prix d’une désaturation artérielle. Le gain tient à l’équilibre obtenu entre l’amélioration du transport d’O2 d’une part, et la désaturation artérielle due au shunt D-G de l’autre [1].
| Hypertension artérielle pulmonaire (HTAP) des congénitaux |
|
Définition de l’HTAP : PAPmoy > 25 mmHg au repos. Trois mécanismes principaux:
- Hypertension passive sur dysfonction du cœur gauche
- Hypertension résistive sur élévation des RAP
- Hypertension hyperkinétique sur élévation du flux (shunt G-D)
Syndrome d'Eisenmenger (HTAP irréversible): PAP moy > 50 mmHg, RAP > 800 dynes•cm•s-5 et shunt bidirectionnel avec cyanose (shunt G-D + composante D-G)
Critère d’inopérabilité: rapport RAP/RAS > 0.7
Caractéristiques hémodynamiques
- Hypotension systémique: aggravation de la composante D-G du shunt
- Débit pulmonaire bas et fixe, hypoxémie à l'effort ou lors d’hypovolémie
- Réactivité vasculaire pulmonaire conservée
- HVD: débit du VD dépendant de sa précharge, intolérance à l'hypovolémie
- Hypotension systémique: risque ischémique du VD, déséquilibre de l'interdépendance ventriculaire
|
| raitement de l’hypertension pulmonaire |
|
Correction de l’acidose, de l’hypothermie, du stress et de la douleur
Ventilation et inhalation (pas d’effet systémique)
- Hyperventilation normobarique
- NO• (10-30 ppm)
- Nébulisation de prostacycline (iloprost), de milrinone ou de nitroglycérine
Vasodilatateurs pulmonaires (risque d’hypotension systémique)
- Prostacyclines (époprosténol, treprostinil)
- Inhibiteurs des phosphodiestérases-5 (sildenafil)
- Inhibiteurs du récepteur de l’endothéline (bosentan)
- Inhibiteurs des phosphodiestérases-3 (milrinone)
- Cardiostimulants: milrinone + adrenaline, dobutamine, levosimendan
- Peu efficaces: nitroglycérine, nesiritide, riociguat, Mg2+
- Anticalciques contre-indiqués chez les congénitaux
Vasoconstricteurs systémiques
- Noradrénaline
- Vasopressine
Procédés non-pharmacologiques
- ECMO, assistance ventriculaire droite
- Septostomie interauriculaire
- Thrombendarterectomie pulmonaire post-embolie
- Transplantation pulmonaire ou cardio-pulmonaire
La clef de l’amélioration hémodynamique n’est pas seulement une baisse de la PAP, mais une baisse de la PVC et une augmentation du volume systolique, traduisant l’amélioration de la fonction du VD
|
La ventilation est un compromis entre une hyperventilation active et le maintien d'une pression intrathoracique moyenne (Pit) basse. Si le volume courant est faible, on risque des atélectasies, une hypercarbie, et une augmentation des RAP dans les petits vaisseaux péri-alvéolaires; s'il est élevé, l'hyperinflation augmente la Pit et comprime les gros vaisseaux extra-alvéolaires [15]. Le volume courant idéal correspond à celui de la capacité résiduelle fonctionnelle (CRF) (Figure 15.68).

Figure 15.68 : Evolution des résistances artérielles pulmonaires (RAP) avec la ventilation. A : Situation normale. A bas volume courant, les RAP s'élèvent dans les petits vaisseaux par hypoventilation et hypercapnie. A haut volume courant, elles s'élèvent dans les gros vaisseaux périalvéolaires qui sont étirés et comprimés mécaniquement. La résultante (courbe rouge) montre que le meilleur compromis est obtenu au volume de la capacité résiduelle fonctionnelle (CRF). Ceci correspond à une ventilation à 6-8 ml/kg [15]. B : En cas d’hypertension pulmonaire chronique, les parois artérielles sont épaissies, avec une média muscularisée, une prolifération sous-endothéliale et une fibrose de l’externa (coupe d’une artère pulmonaire en cartouche). Ces vaiseaux ne sont plus compressibles par la pression intrathoracique. La courbe corrrespondant aux gros vaisseaux est donc aplatie, et les RAP totales n’ont plus une forme en "U" ; elles n’augmentent plus à haut volume courant. Ces patients tolèrent donc très bien l’hyperventilation en IPPV.
Il faut jouer sur le volume courant, la fréquence et le mode ventilatoire (mode en pression-contrôlée) pour obtenir la PaCO2 et la Pit moy les plus basses possible. De ce point de vue, la durée de l'inspirium augmente davantage la Pit moy que la valeur du pic inspiratoire de pression [28]. Toutefois, la partie droite du graphique de la Figure 15.67 ne s’applique pas réellement au patient souffrant d’HTAP, parce que la paroi épaissie et rigide de ses vaisseaux pulmonaires empêche toute compression par une ventilation à haut volume courant. Il n’y a donc pas lieu de craindre une augmentation supplémentaire de la PAP lors d’une hyperventilation mécanique.
Souvent, on préfère proposer une anesthésie loco-régionale par crainte des interférences ventilatoires d'une anesthésie générale. Dans le cas présent, cette crainte est infondée. En effet, l'accroissement de postcharge pour le VD que représente l'IPPV est très faible par rapport à sa postcharge habituelle: ajouter une Pit moy de 10 mmHg à une PAPmoy de 50 mmHg modifie moins les conditions hémodynamiques que lorsque la PAPmoy est normale (≤ 15 mmHg). De plus, l'IPPV offre la possibilité d'hyperventiler le patient et, ce faisant, de diminuer ses RAP. A l'inverse, la vasodilatation artérielle d'une anesthésie loco-régionale rachidienne risque d'aggraver la composante D-G du shunt, de provoquer une désaturation artérielle et d'accentuer la cyanose [2]. La réaction hémodynamique du patient à l'IPPV peut être testée en préopératoire en lui faisant réaliser une manoeuvre de Valsalva une fois le cathéter artériel en place et en observant l'évolution de la pression artérielle. Le plus souvent, les variations respiratoires sont atténuées et la pression moyenne (PAM) est stabilisée à une valeur très légèrement inférieure (< 10%) à sa valeur en spontanée. Ceci laisse présager une bonne tolérance à l’IPPV.
Anesthésie et hypertension pulmonaire
A l'exception de la kétamine, du N2O et du desflurane qui provoquent une stimulation sympathique et une augmentation des RAP chez l’adulte [12,14], les agents d'anesthésie ont peu d'influence sur la circulation pulmonaire. Les recommandations pour l’anesthésie du patient souffrant d’HTAP peuvent se formuler de la manière suivante [11,15,16,27,28]:
- Anesthésie profonde (stress-free) à base d’un fentanil et d’un agent hypnogène (halogéné, propofol);
- Hyperventilation normobarique (IPPV), alcalose respiratoire; utilisation du NO (10-20 ppm);
- Normothermie;
- Maintien de la précharge : éviter toute hypovolémie;
- Maintien des RAS (vasoconstricteur): pression de perfusion coronarienne, diminution de la composante D-G du shunt, repositionnement du septum interventriculaire.
- Monitorage: un cathéter artériel et une PVC sont requis. Un système de mesure du débit cardiaque est nécessaire pour les interventions majeures, mais le cathéter pulmonaire peut être difficile à placer ou même dangereux selon les configurations anatomiques; un dispositif de type PiCCO est très utile.
- Seuil de transfusion: Hb 100 g/L.
L’anesthésie générale avec hyperventilation mécanique remplit mieux ces critères qu’une anesthésie loco-régionale (ALR) rachidienne. En effet, cette dernière diminue les RAS et la précharge droite, ce qui augmente la composante D-G du shunt et limite le débit pulmonaire. Ceci est particulièrement marqué pour la rachi-anesthésie; lorsqu’elle est requise, la péridurale est possible à condition que le bloc soit installé très lentement et que ses effets hémodynamiques soient simultanément compensés. D’une manière générale, l’ALR est donc plutôt mal adaptée à l’HTAP.
| Anesthésie et HTAP |
|
Eviter toute augmentation des RAP : hypoventilation, hypercarbie, atélectasies, surpression endothoracique, acidose, hypothermie, stimulation sympathique, stress, douleur
Anesthésie: - Anesthésie et analgésie profondes
- Hyperventilation normobarique, alcalose respiratoire, normoxie, NO
- Maintien de la précharge
- Maintien des RAS
- Normothermie
- Rachianesthésie mal tolérée, péridurale possible si bloc d’installation très lente
L’IPPV est bien tolérée pour 3 raisons
- L’augmentation de postcharge droite est faible par rapport à la PAP chroniquement haute
- Le VD hypertrophié supporte une augmentation de postcharge
- Les vaisseaux pulmonaires épaissis et fibrosés ne sont pas compressibles par l’IPPV
|
© BETTEX D, CHASSOT PG, Janvier 2008, dernière mise à jour Janvier 2018
Références
- ALTHOFF TF, KNEBEL P, PANDA A, et al. Long-term follow-up of a fenestrated Amplatzer atrial septal occluder in pulmonary arterial hypertemsion. Chest 2008; 133:283-5
- AMMASH NA, CONNOLLY HM, ABEL MD, WARNES CA. Noncardiac surgery in Eisenmenger syndrome. J Am Coll Cardiol 1999; 33:222-7
- BARR FE, MACRAE D, Inhaled nitric oxide and related therapies. Pediatr Crit Care Med 2010; 11(Suppl): S30-S36
- BAUMGARTNER H, BONHOEFFER P, DE GROOT NMS, et al. ESC Guidelines for the management of grown-up congenital heart disease (new version 2010). Eur Heart J 2010; 31:2915-57
- BELZER DT, KORT HW, DAY RW, et al. Inhaled nitric oxide as a preoperative test (INOP Test I): the INOP Test Study Group. Circulation 2002; 106(Suppl I):S76-81
- BENZA RL, MILLER DP, BARST RJ, et al. An evaluation of long-term survival from time of diagnosis in pulmonary arterial hypertension from REVEAL. Chest 2012; 142:448-56
- BLAISE G, LANGLEBEN D, HUBERT B. Pulmonary arterial hypertension. Pathophysiology and anesthetic approach. Anesthesiology 2003; 99:1415-32
- BRONICKI RA, BADEN HP. Pathophysiology of right ventricular failure in pulmonary hypertension. Pediatr Crit Care Med 2010; 11(Suppl):S15-S22
- BUNDTS W, VAN PELT N, GILLYNS H, et al. Residual pulmonary vasoreactivity to inhaled nitric oxide in patients with severe obstructive pulmonary hypertension and Eisenmenger syndrome. Heart 2001; 86:553-9
- CANNESSON M, EARING MG, COLLANGE V, KERSTEN JR. Anesthesia for noncardiac surgery in adults with congenital heart disease. Anesthesiology 2009; 111:432-40
- CHASSOT PG, BETTEX DA. Anesthesia and adult congenital heart disease. J Cardiothorac Vasc Anesth 2006; 20:414-37
- CIOFOLO M, REIZ S. Circulatory effects of volatile anesthetic agents. Minerva Anaesthesiol 1999; 65:232-8
- DALIENTO L, SOMERVILLE J, PRESBITERO P, et al. Eisenmenger syndrome. Factors relating to deterioration and death. Eur Heart J 1998; 19:1845-55
- EBERT TJ, MUZI M. Sympathetic hyperactivity during desflurane anesthesia in healthy volunteers: a comparison with isoflurane. Anesthesiology 1993; 79:444-53
- FISCHER LG, VAN HAKEN H, BÜRKLE H. Management of pulmonary hypertension: Physiological and pharmacological considerations for anesthesiologists. Anesth Analg 2003; 96:1603-1664 FRANK SM, FLEISHER LA, BRESLOW MJ, et al. Perioperative maintenance of normothermia reduces the incidence of morbid cardiac events. A randomized clinical trial. JAMA 1997; 277: 1127-34
- FRANK SM, FLEISHER LA, BRESLOW MJ, et al. Perioperative maintenance of normothermia reduces the incidence of morbid cardiac events. A randomized clinical trial. JAMA 1997; 277: 1127-34
- GALIÉ N, HUMBERT M, VACHIERY JL, et al. 2015 ESC/ERS Guideline for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2016; 37:67-119
- GALLE N, HOEPER MM, HUMBERT M, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2009; 30:2493-537
- GHOFRANI HA, WIEDEMAN R, ROSE F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension. A randomized controlled trial, Lancet 2002; 360:895-900
- GUAZZI M, BORLAUG BA. Pulmonary hypertension due to left heart disease. Circulation 2012; 126:975-90
- HADDAD F, COUTURE P, TOUSIGNANT C, DENAULT AY. The right ventricle in cardiac surgery, a perioperative perspective: I. Anatomy, physiology and assessment. Anesth Analg 2009; 108:407-21
- HADDAD F, DOYLE R, MURPHY DJ. HUNT SA. Right ventricular function in cardiovascular disease, Part II. Pathophysiology, clinical importance and management of right ventricular failure. Circulation 2008; 117:1717-31
- HOEPER MM, BOGAARD HJ, CONDLIFFE R, et al. Definitions and diagnosis of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D42-50
- HOPKINS WE, OCHOA LL, RICHARDSON GW, TRULOCK EP. Comparison of the hemodynamics and survival of adults with severe primary pulmonary hypertension or Eisenmenger syndrome. J Heart Lung Transplant 1996; 15:100-5
- HUMBERT M, SITBON O, SIMONEAU G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004; 351:1425-36
- JEON Y, RYU JH, LIM YJ, et al. Comparative hemodynamic effects of vasopressin and norepinephrine after milrinone-induced hypotension in off-pump coronary artery bypass surgical patients. Eur J Cardiothorac Surg 2006; 29:952-6
- KERBAUL F, RONDELET B, COLLART F, et al. Hypertension artérielle pulmonaire en anesthésie-réanimation. Ann Fr Anesth Réanim 2005; 24:528-40
- LOVELL AT. Anaesthetic implications of grown-up congenital heart disease. Br J Anaesth 2004; 93:129-39
- MICHELAKIS E, TYMCHAK W, LIEN D, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: Comparison with inhaled nitric oxide. Circulation 2002; 105:2398-403
- OECHSLIN E. Eisenmenger's syndrome. In: GATZOULIS MA, WEBB GD, DAUBENEY PEF, Eds. Diagnosis and management of adult congenital heart disease. Edinburgh: Churchill Livingstone 2003, 363-77
- OPOTOWSKY AR. Clinical evaluation and management of pulmonary hypertension in the adult with congenital heart disease. Circulation 2015; 131:200-10
- PALEVSKY HI, SCHLOO BL, PIETRA CC, et al. Primary pulmonary hypertension: Vascular structure, morphometry, and responsiveness to vasodilator agents. Circulation 1989; 80:1207-21
- POLLESELLO P, PARISSIS J, KIVIKKO M, et al. Levosimendan meta-analyses: is there a pattern in the effect on mortality ? Int J Cardiol 2016; 209: 77-83
- RABINOVITCH M, HAWORTH SG, CASTANEDA AR, et al. Lung biopsy in congenital heart disease: a morphometric approach to pulmonary vascular disease. Circulation 1978; 58:1007-22
- RICH S, MCLAUGHLIN VV. The effects of chronic prostacyclin therapy on cardiac output and symptoms in primary pulomonary hypertension. J Am Coll Cardiol 1999; 34:1184-7
- SCHROEDER RA, WOOD GL, PLOTKIN JS, et al. Intraoperative use of inhaled PGI(2) for acute pulmonary hypertension and right ventricular failure. Anesth Analg 2000; 91:291-5
- SIMONEAU G, GATZOULIS MA, ADAIA I,, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013; 62:D34-41
- THUNBERG CA, GAITAN BD, GREWAL A, et al. Pulmonary hypertension in patients undergoing cardiac surgery: pathophysiology, perioperative management and outcome. J Cardiothorac Vasc Anesth 2013; 27: 551-72
- WINTERHALTER M, ANTONIOU T, LOUKANOV T. Management of adult patients with perioperative pulmonary hypertension: technical aspects and therapeutic options. Cardiology 2010; 116:3-9